Tricor 160mg, 200mg Fenofibrate Zastosowanie, skutki uboczne i dawkowanie. Cena w aptece internetowej. Leki generyczne bez recepty.
Co to jest Tricor i jak się go stosuje?
Tricor to lek na receptę stosowany w celu zmniejszenia objawów cholesterolu i trójglicerydów (kwasów tłuszczowych) we krwi. Tricor może być stosowany samodzielnie lub z innymi lekami.
- Tricor należy do klasy leków zwanych Fibric Acid Agents.
- Nie wiadomo, czy Tricor 200mg jest bezpieczny i skuteczny u dzieci
Jakie są możliwe skutki uboczne Tricora?
Tricor 200mg może powodować poważne skutki uboczne, w tym:
- ostry ból brzucha rozprzestrzeniający się na plecy lub łopatkę,
- utrata apetytu,
- ból brzucha po samym posiłku,
- zażółcenie skóry lub oczu (żółtaczka),
- gorączka,
- dreszcze,
- słabość,
- ból gardła,
- owrzodzenia jamy ustnej,
- nietypowe siniaki lub krwawienia,
- ból w klatce piersiowej,
- nagły kaszel,
- świszczący oddech,
- szybkie oddychanie,
- kaszel krwią i
- obrzęk, ciepło lub zaczerwienienie ręki lub nogi
Natychmiast uzyskaj pomoc medyczną, jeśli wystąpi którykolwiek z wymienionych powyżej objawów.
Najczęstsze skutki uboczne Tricor to:
- katar,
- kichanie i
- nieprawidłowe testy laboratoryjne
Poinformuj lekarza, jeśli wystąpią jakiekolwiek skutki uboczne, które Ci przeszkadzają lub które nie ustępują.
To nie wszystkie możliwe skutki uboczne Tricora. Aby uzyskać więcej informacji, skontaktuj się z lekarzem lub farmaceutą.
Zadzwoń do lekarza, aby uzyskać poradę medyczną na temat skutków ubocznych. Możesz zgłosić skutki uboczne do FDA pod numerem 1-800-FDA-1088.
OPIS
TRICOR (tabletki fenofibratu) to środek regulujący lipidy dostępny w postaci tabletek do podawania doustnego. Każda tabletka zawiera 54 mg lub 160 mg fenofibratu. Nazwa chemiczna fenofibratu to ester 1-metyloetylowy kwasu 2-[4-(4-chlorobenzoilo)fenoksy]-2-metylopropanowego o następującym wzorze strukturalnym:
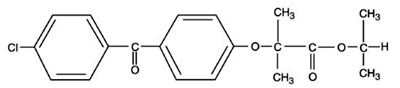
Wzór empiryczny to C20H21O4Cl, a masa cząsteczkowa to 360,83; fenofibrat jest nierozpuszczalny w wodzie. Temperatura topnienia 79-82°C. Fenofibrat jest białym ciałem stałym, stabilnym w normalnych warunkach.
Nieaktywne składniki
Każda tabletka zawiera koloidalny dwutlenek krzemu, krospowidon, monohydrat laktozy, lecytynę, celulozę mikrokrystaliczną, alkohol poliwinylowy, powidon, laurylosiarczan sodu, stearylofumaran sodu, talk, dwutlenek tytanu i gumę ksantanową. Ponadto pojedyncze tabletki 54 mg zawierają D&C Yellow nr 10, FD&C Yellow nr 6, FD&C Blue nr 2.
WSKAZANIA
Pierwotna hipercholesterolemia lub mieszana dyslipidemia
TRICOR jest wskazany jako terapia wspomagająca dietę w celu obniżenia podwyższonego cholesterolu lipoprotein o małej gęstości (LDL-C), cholesterolu całkowitego (Całkowitego), trójglicerydów i apolipoproteiny B (Apo B) oraz w celu zwiększenia stężenia cholesterolu lipoprotein o dużej gęstości (HDL- C) u dorosłych pacjentów z pierwotną hipercholesterolemią lub mieszaną dyslipidemią.
Ciężka hipertriglicerydemia
TRICOR 200mg jest również wskazany jako terapia wspomagająca dietę w leczeniu dorosłych pacjentów z ciężką hipertriglicerydemią. Poprawa kontroli glikemii u pacjentów z cukrzycą wykazujących chylomikronemię na czczo zwykle pozwala uniknąć konieczności interwencji farmakologicznej.
Znacznie podwyższone poziomy triglicerydów w surowicy (np. > 2000 mg/dl) mogą zwiększać ryzyko rozwoju zapalenia trzustki. Wpływ terapii fenofibratem na zmniejszenie tego ryzyka nie został odpowiednio zbadany.
Ważne ograniczenia użytkowania
W dużym, randomizowanym badaniu klinicznym z udziałem pacjentów z cukrzycą typu 2 nie wykazano, że fenofibrat w dawce odpowiadającej 145 mg preparatu TRICOR zmniejsza zachorowalność i śmiertelność z powodu choroby wieńcowej serca [patrz OSTRZEŻENIA I ŚRODKI ].
DAWKOWANIE I SPOSÓB PODAWANIA
Uwagi ogólne
Pacjenci powinni być umieszczeni na odpowiedniej diecie hipolipemizującej przed otrzymaniem preparatu TRICOR 160 mg i powinni kontynuować tę dietę podczas leczenia preparatem TRICOR. Tabletki TRICOR można podawać niezależnie od posiłków.
Początkowym leczeniem dyslipidemii jest terapia dietetyczna specyficzna dla typu nieprawidłowości lipoprotein. Nadmierna masa ciała i nadmierne spożycie alkoholu mogą być ważnymi czynnikami hipertriglicerydemii i należy je rozwiązać przed rozpoczęciem jakiejkolwiek terapii lekowej. Ćwiczenia fizyczne mogą być ważnym środkiem pomocniczym. Choroby przyczyniające się do hiperlipidemii, takie jak niedoczynność tarczycy lub cukrzyca, powinny być poszukiwane i odpowiednio leczone. Terapia estrogenami, diuretykami tiazydowymi i beta-blokerami czasami wiąże się z ogromnym wzrostem stężenia triglicerydów w osoczu, zwłaszcza u osób z hipertriglicerydemią rodzinną. W takich przypadkach odstawienie specyficznego czynnika etiologicznego może wyeliminować potrzebę specyficznej terapii farmakologicznej hipertriglicerydemii.
Poziomy lipidów należy okresowo monitorować i należy rozważyć zmniejszenie dawki preparatu TRICOR, jeśli poziom lipidów spadnie znacznie poniżej docelowego zakresu.
Terapię należy przerwać u pacjentów, którzy nie wykazują odpowiedniej odpowiedzi po dwóch miesiącach leczenia maksymalną zalecaną dawką 145 mg raz na dobę.
Pierwotna hipercholesterolemia lub mieszana dyslipidemia
Początkowa dawka leku TRICOR 200 mg wynosi 145 mg raz na dobę.
Ciężka hipertriglicerydemia
Dawka początkowa wynosi od 48 do 145 mg na dobę. Dawkowanie należy dostosować indywidualnie do odpowiedzi pacjenta i w razie potrzeby dostosować po powtórnym oznaczaniu stężenia lipidów w odstępach 4–8 tygodni. Maksymalna dawka to 145 mg raz na dobę.
Upośledzona funkcja nerek
Leczenie preparatem TRICOR 200 mg należy rozpoczynać od dawki 48 mg na dobę u pacjentów z łagodnymi do umiarkowanych zaburzeniami czynności nerek i zwiększać dopiero po ocenie wpływu tej dawki na czynność nerek i stężenie lipidów. Należy unikać stosowania preparatu TRICOR u pacjentów z ciężkimi zaburzeniami czynności nerek [patrz Używaj w określonych populacjach oraz FARMAKOLOGIA KLINICZNA ].
Pacjenci geriatryczni
Doboru dawki u osób w podeszłym wieku należy dokonać na podstawie czynności nerek [patrz Używaj w określonych populacjach ].
JAK DOSTARCZONE
Formy dawkowania i mocne strony
- 48 mg żółte tabletki, z nadrukowanymi literami identyfikacyjnymi kodu „FI”.
- 48 mg żółte tabletki z nadrukiem logo „a” i literami identyfikacyjnymi kodu „FI”.
- Białe tabletki 145 mg, z nadrukowanymi literami identyfikacyjnymi kodu „FO”.
- Białe tabletki 145 mg, z nadrukiem „a” i literami identyfikacyjnymi kodu „FO”.
Składowania i stosowania
TRICOR® (tabletki fenofibratu) jest dostępny w dwóch mocach:
48 mg
Żółte tabletki z nadrukiem z literami identyfikacyjnymi kodu „FI”, dostępne w butelkach po 90 ( NDC 0074-3173-90).
Tabletki żółte, z nadrukiem logo „a” i literami identyfikacyjnymi kodu „FI”, dostępne w butelkach po 90 ( NDC 0074-6122-90).
145 mg
Białe tabletki z nadrukiem literami identyfikacyjnymi kodu „FO”, dostępne w butelkach po 90 ( NDC 0074-3189-90).
Białe tabletki z nadrukiem logo „a” i literami identyfikacyjnymi kodu „FO”, dostępne w butelkach po 90 ( NDC 0074-6123-90).
Magazynowanie
Przechowywać w 25°C (77°F); wycieczki dozwolone do 15-30 ° C (59-86 ° F).
[Patrz Kontrolowana temperatura pomieszczenia USP]. Trzymać z dala od dzieci. Chronić przed wilgocią.
Wyprodukowano dla AbbVie Inc., North Chicago, IL 60064, USA przez Fournier Laboratories Ireland Limited, Anngrove, Carrigtwohill Co. Cork, Irlandia. Poprawiono : marzec 2021
SKUTKI UBOCZNE
Poniżej oraz w innych miejscach na etykiecie opisano następujące poważne działania niepożądane:
- Śmiertelność i zachorowalność na chorobę wieńcową [patrz OSTRZEŻENIA I ŚRODKI ]
- Hepatotoksyczność [patrz OSTRZEŻENIA I ŚRODKI ]
- Zapalenie trzustki [patrz OSTRZEŻENIA I ŚRODKI ]
- Reakcje nadwrażliwości [patrz OSTRZEŻENIA I ŚRODKI ]
- Choroba żylno-zakrzepowo-zatorowa [patrz OSTRZEŻENIA I ŚRODKI ]
Doświadczenie w badaniach klinicznych
Ponieważ badania kliniczne prowadzone są w bardzo zróżnicowanych warunkach, częstość występowania działań niepożądanych obserwowanych w badaniach klinicznych leku nie może być bezpośrednio porównywana z częstością w badaniach klinicznych innego leku i może nie odzwierciedlać częstości obserwowanych w praktyce.
Zdarzenia niepożądane zgłaszane przez 2% lub więcej pacjentów leczonych fenofibratem (i więcej niż placebo) podczas badań z podwójnie ślepą próbą, kontrolowanych placebo, niezależnie od przyczyn, wymieniono w Tabeli 1 poniżej. Zdarzenia niepożądane doprowadziły do przerwania leczenia u 5,0% pacjentów leczonych fenofibratem iu 3,0% leczonych placebo. W badaniach z podwójnie ślepą próbą najczęstszymi zdarzeniami były zwiększenie wyników testów czynnościowych wątroby, które spowodowały przerwanie leczenia fenofibratem u 1,6% pacjentów.
W kontrolowanych badaniach pokrzywkę obserwowano u 1,1% w porównaniu do 0%, a wysypkę u 1,4% w porównaniu z 0,8% pacjentów otrzymujących fenofibrat i placebo, odpowiednio.
Wzrost enzymów wątrobowych
W zbiorczej analizie 10 badań kontrolowanych placebo, zwiększenie aktywności AlAT do >3-krotności górnej granicy normy wystąpiło u 5,3% pacjentów przyjmujących fenofibrat w dawkach równoważnych 96 mg do 145 mg TRICOR na dobę w porównaniu z 1,1% pacjentów otrzymujących placebo [Widzieć OSTRZEŻENIA I ŚRODKI ]. W 8-tygodniowym badaniu częstość występowania zwiększenia aktywności AlAT lub AspAT ≥ 3-krotności górnej granicy normy wynosiła 13% u pacjentów otrzymujących dawki odpowiadające 96 mg do 145 mg TRICOR na dobę i wynosiła 0% u pacjentów otrzymujących dawki odpowiadające 48 mg lub mniej TRICOR dziennie lub placebo.
Doświadczenie postmarketingowe
Następujące działania niepożądane zostały zidentyfikowane podczas stosowania fenofibratu po dopuszczeniu do obrotu. Ponieważ reakcje te są zgłaszane dobrowolnie w populacji o niepewnej wielkości, nie zawsze jest możliwe wiarygodne oszacowanie ich częstości lub ustalenie związku przyczynowego z ekspozycją na lek: bóle mięśni, rabdomioliza, zapalenie trzustki, ostra niewydolność nerek, skurcze mięśni, zapalenie wątroby, marskość wątroby, bilirubina całkowita, niedokrwistość, bóle stawów, zmniejszenie stężenia hemoglobiny, zmniejszenie hematokrytu, zmniejszenie liczby białych krwinek, osłabienie, poważnie obniżony poziom cholesterolu HDL i śródmiąższowa choroba płuc. Reakcje nadwrażliwości na światło występowały kilka dni lub miesięcy po rozpoczęciu; w niektórych z tych przypadków pacjenci zgłaszali wcześniejszą reakcję nadwrażliwości na światło na ketoprofen.
INTERAKCJE Z LEKAMI
Antykoagulanty kumaryny
Obserwowano nasilenie działania przeciwzakrzepowego typu kumaryny wraz z wydłużeniem PT/INR.
Należy zachować ostrożność, gdy leki przeciwzakrzepowe kumaryny są podawane w połączeniu z produktem TRICOR. Dawkę antykoagulantów należy zmniejszyć, aby utrzymać PT/INR na pożądanym poziomie, aby zapobiec powikłaniom krwotocznym. Częste oznaczanie PT/INR jest wskazane, dopóki nie zostanie definitywnie ustalone, że PT/INR ustabilizowało się [patrz OSTRZEŻENIA I ŚRODKI ].
Leki immunosupresyjne
Leki immunosupresyjne, takie jak cyklosporyna i takrolimus, mogą powodować nefrotoksyczność ze zmniejszeniem klirensu kreatyniny i wzrostem stężenia kreatyniny w surowicy, a ponieważ wydalanie nerkowe jest główną drogą eliminacji fibratów, w tym TRICOR, istnieje ryzyko, że interakcja doprowadzi do pogorszenia czynności nerek. Należy dokładnie rozważyć korzyści i ryzyko związane ze stosowaniem preparatu TRICOR (tabletki fenofibratu) z lekami immunosupresyjnymi i innymi potencjalnie nefrotoksycznymi środkami, a także monitorować najniższą skuteczną dawkę i czynność nerek.
Żywice wiążące kwasy żółciowe
Ponieważ żywice wiążące kwasy żółciowe mogą wiązać inne leki podawane jednocześnie, pacjenci powinni przyjmować TRICOR 200 mg co najmniej 1 godzinę przed lub 4 do 6 godzin po żywicy wiążącej kwasy żółciowe, aby uniknąć utrudniania jej wchłaniania.
Kolchicyna
Przypadki miopatii, w tym rabdomiolizy, zgłaszano podczas jednoczesnego podawania fenofibratów z kolchicyną, dlatego należy zachować ostrożność przepisując fenofibrat z kolchicyną.
OSTRZEŻENIA
Zawarte jako część ŚRODKI OSTROŻNOŚCI Sekcja.
ŚRODKI OSTROŻNOŚCI
Śmiertelność i zachorowalność na chorobę wieńcową
Nie ustalono wpływu produktu TRICOR 160 mg na zachorowalność i śmiertelność z powodu choroby niedokrwiennej serca oraz śmiertelność z przyczyn innych niż sercowo-naczyniowe.
Badanie Action to Control Cardiovascular Risk in Diabetes Lipid (ACCORD Lipid) było randomizowanym, kontrolowanym placebo badaniem obejmującym 5518 pacjentów z cukrzycą typu 2, leczonych fenofibratem jako podstawową terapię statynami. Średni czas obserwacji wyniósł 4,7 roku. Terapia skojarzona fenofibratem i statynami wykazała nieistotne, 8% zmniejszenie względnego ryzyka wystąpienia pierwszorzędowego punktu końcowego poważnych niepożądanych zdarzeń sercowo-naczyniowych (MACE), złożonego z niezakończonych zgonem zawału mięśnia sercowego, udaru mózgu niezakończonego zgonem i zgonu z powodu choroby sercowo-naczyniowej (współczynnik ryzyka) HR] 0,92, 95% CI 0,79-1,08 (p=0,32) w porównaniu do monoterapii statynami. W analizie podgrup płci współczynnik ryzyka dla MACE u mężczyzn otrzymujących terapię skojarzoną w porównaniu z monoterapią statyną wyniósł 0,82 (95% CI 0,69-0,99), a współczynnik ryzyka dla MACE u kobiet otrzymujących terapię skojarzoną w porównaniu z monoterapią statyną wyniósł 1,38 (95% CI 0,98-1,94 (interakcja p=0,01). Kliniczne znaczenie odkrycia tej podgrupy jest niejasne.
Badanie Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) było 5-letnim randomizowanym, kontrolowanym placebo badaniem obejmującym 9795 pacjentów z cukrzycą typu 2 leczonych fenofibratem. Fenofibrat wykazał nieistotne 11% względne zmniejszenie pierwszorzędowego punktu końcowego zdarzeń choroby wieńcowej (współczynnik ryzyka [HR] 0,89, 95% CI 0,75-1,05, p=0,16) oraz znamienne 11% zmniejszenie drugorzędowego wyniku całkowitego zdarzenia sercowo-naczyniowe (HR 0,89 [0,80-0,99], p=0,04). Odnotowano nieistotny 11% (HR 1,11 [0,95, 1,29], p=0,18) i 19% (HR 1,19 [0,90, 1,57], p=0,22) wzrost śmiertelności, odpowiednio, całkowitej i z powodu choroby wieńcowej serca po zastosowaniu fenofibratu w porównaniu z placebo.
Ze względu na podobieństwa chemiczne, farmakologiczne i kliniczne między preparatem TRICOR (tabletki fenofibratu), klofibratem i gemfibrozylem, niekorzystne wyniki 4 dużych randomizowanych, kontrolowanych placebo badań klinicznych z tymi innymi lekami fibratowymi mogą również dotyczyć preparatu TRICOR.
Coronary Drug Project, dużym badaniu pacjentów po zawale mięśnia sercowego leczonych przez 5 lat klofibratem, nie zaobserwowano różnicy w śmiertelności między grupą klofibratu a grupą placebo. Zaobserwowano jednak różnicę w częstości występowania kamicy żółciowej i zapalenia pęcherzyka żółciowego wymagającego operacji między obiema grupami (3,0% vs 1,8%).
badaniu przeprowadzonym przez Światową Organizację Zdrowia (WHO) 5000 pacjentów bez rozpoznanej choroby wieńcowej było leczonych placebo lub klofibratem przez 5 lat i obserwowanych przez dodatkowy rok. W grupie klofibratu w porównaniu z grupą placebo stwierdzono znamienną statystycznie wyższą śmiertelność całkowitą, skorygowaną względem wieku (5,70% vs. 3,96%, p =
Helsinki Heart Study było dużym (n=4081) badaniem obejmującym mężczyzn w średnim wieku bez choroby wieńcowej w wywiadzie. Badani otrzymywali albo placebo albo gemfibrozil przez 5 lat, z 3,5 letnim otwartym przedłużeniem później. Całkowita śmiertelność była liczbowo wyższa w grupie randomizowanej gemfibrozylem, ale nie osiągnęła istotności statystycznej (p = 0,19, 95% przedział ufności dla ryzyka względnego G:P = 0,91-1,64). Chociaż śmiertelność z powodu raka wykazywała wyższą tendencję w grupie gemfibrozylu (p = 0,11), nowotwory (z wyjątkiem raka podstawnokomórkowego) były diagnozowane z równą częstością w obu grupach badawczych. Ze względu na ograniczoną wielkość badania, względne ryzyko zgonu z jakiejkolwiek przyczyny nie różniło się od obserwowanego w danych z 9-letniego okresu obserwacji z badania Światowej Organizacji Zdrowia (RR=1,29).
Do składowej prewencji wtórnej Helsinki Heart Study włączono mężczyzn w średnim wieku wykluczonych z badania prewencji pierwotnej z powodu rozpoznanej lub podejrzewanej choroby wieńcowej serca. Badani otrzymywali gemfibrozil lub placebo przez 5 lat. Chociaż zgony sercowe wykazywały wyższą tendencję w grupie gemfibrozylu, nie było to statystycznie istotne (współczynnik ryzyka 2,2, 95% przedział ufności: 0,94-5,05). Częstość operacji pęcherzyka żółciowego nie była statystycznie istotna między grupami badanymi, ale wykazywała tendencję wyższą w grupie gemfibrozylu (1,9% vs 0,3%, p = 0,07).
Hepatotoksyczność
Po wprowadzeniu produktu TRICOR do obrotu zgłaszano poważne polekowe uszkodzenie wątroby (DILI), w tym przeszczepienie wątroby i zgon. DILI zgłaszano w ciągu pierwszych kilku tygodni leczenia lub po kilku miesiącach leczenia, aw niektórych przypadkach ustępowało po przerwaniu leczenia TRICOR 200 mg. U pacjentów z DILI występowały objawy przedmiotowe i podmiotowe, w tym ciemny mocz, nieprawidłowy stolec, żółtaczka, złe samopoczucie, ból brzucha, ból mięśni, utrata masy ciała, świąd i nudności. Wielu pacjentów miało jednocześnie podwyższone stężenie bilirubiny całkowitej, transaminazy alaninowej (ALT) i transaminazy asparaginianowej (AST). DILI scharakteryzowano jako wątrobowokomórkowe, przewlekłe aktywne i cholestatyczne zapalenie wątroby, a marskość występuje w połączeniu z przewlekłym aktywnym zapaleniem wątroby.
W badaniach klinicznych stosowanie fenofibratu w dawkach równoważnych 96 mg do 145 mg TRICOR 160 mg na dobę wiązało się ze zwiększeniem aktywności AspAT lub AlAT w surowicy. Częstość występowania zwiększenia aktywności aminotransferaz może być zależna od dawki [patrz DZIAŁANIA NIEPOŻĄDANE ].
TRICOR 160 mg jest przeciwwskazany u pacjentów z czynną chorobą wątroby, w tym z pierwotną marskością żółciową wątroby i niewyjaśnionymi uporczywymi zaburzeniami czynności wątroby [patrz PRZECIWWSKAZANIA ]. Monitoruj czynność wątroby pacjenta, w tym ALT, AspAT i bilirubinę całkowitą w surowicy przed rozpoczęciem leczenia oraz okresowo przez cały czas trwania leczenia preparatem TRICOR. Przerwać podawanie TRICOR 200 mg, jeśli pojawią się oznaki lub objawy uszkodzenia wątroby lub jeśli podwyższony poziom enzymów utrzymuje się (AlAT lub AspAT > 3 razy powyżej górnej granicy normy lub jeśli towarzyszy mu podwyższenie stężenia bilirubiny). Nie należy ponownie rozpoczynać leczenia TRICOR 160mg u tych pacjentów, jeśli nie ma alternatywnego wyjaśnienia uszkodzenia wątroby.
Miopatia i rabdomioliza
Fibraty zwiększają ryzyko miopatii i są związane z rabdomiolizą. Wydaje się, że ryzyko poważnej toksyczności mięśniowej jest zwiększone u pacjentów w podeszłym wieku oraz u pacjentów z cukrzycą, niewydolnością nerek lub niedoczynnością tarczycy.
Miopatię należy rozważyć u każdego pacjenta z rozlanymi bólami mięśniowymi, tkliwością lub osłabieniem mięśni i/lub znacznym podwyższeniem poziomu fosfokinazy kreatynowej (CPK).
Pacjentom należy zalecić, aby niezwłocznie zgłaszali niewyjaśnione bóle mięśni, tkliwość lub osłabienie, szczególnie jeśli towarzyszy im złe samopoczucie lub gorączka. Poziom CPK należy ocenić u pacjentów zgłaszających te objawy, a leczenie produktem TRICOR 200 mg należy przerwać w przypadku wystąpienia znacznie podwyższonego stężenia CPK lub podejrzenia lub rozpoznania miopatii/zapalenia mięśni.
Dane z badań obserwacyjnych wskazują, że ryzyko rabdomiolizy zwiększa się, gdy fibraty, w szczególności gemfibrozyl, są podawane jednocześnie ze statyną. Należy unikać takiego połączenia, chyba że korzyści wynikające z dalszych zmian poziomu lipidów prawdopodobnie przewyższą zwiększone ryzyko tego połączenia leków [patrz FARMAKOLOGIA KLINICZNA ].
Przypadki miopatii, w tym rabdomiolizy, zgłaszano podczas jednoczesnego podawania fenofibratów z kolchicyną i należy zachować ostrożność przepisując fenofibrat z kolchicyną [patrz INTERAKCJE Z LEKAMI ].
Kreatynina w surowicy
U pacjentów leczonych fenofibratem zgłaszano zwiększenie stężenia kreatyniny w surowicy. Te podwyższenia mają tendencję do powrotu do wartości wyjściowych po odstawieniu fenofibratu. Kliniczne znaczenie tych obserwacji nie jest znane. Monitoruj czynność nerek u pacjentów z zaburzeniami czynności nerek przyjmujących TRICOR. Należy również rozważyć monitorowanie czynności nerek u pacjentów przyjmujących TRICOR z ryzykiem niewydolności nerek, takich jak osoby w podeszłym wieku i pacjenci z cukrzycą.
Kamica żółciowa
Fenofibrat, podobnie jak klofibrat i gemfibrozil, może zwiększać wydalanie cholesterolu do żółci, prowadząc do kamicy żółciowej. W przypadku podejrzenia kamicy żółciowej wskazane są badania pęcherzyka żółciowego. Leczenie TRICOR 160 mg należy przerwać w przypadku stwierdzenia kamieni żółciowych.

Antykoagulanty kumaryny
Należy zachować ostrożność, gdy antykoagulanty kumaryny są podawane w skojarzeniu z produktem TRICOR 160 mg ze względu na nasilenie działania przeciwzakrzepowego typu kumaryny w wydłużaniu czasu protrombinowego/międzynarodowego współczynnika znormalizowanego (PT/INR). Aby zapobiec powikłaniom krwotocznym, zaleca się częste monitorowanie PT/INR i dostosowywanie dawki leku przeciwzakrzepowego do czasu ustabilizowania PT/INR [patrz INTERAKCJE Z LEKAMI ].
Zapalenie trzustki
U pacjentów przyjmujących fenofibrat, gemfibrozil i klofibrat zgłaszano zapalenie trzustki. Zjawisko to może oznaczać brak skuteczności u pacjentów z ciężką hipertriglicerydemią, bezpośrednie działanie leku lub wtórne zjawisko związane z tworzeniem się kamieni w drogach żółciowych lub szlamu z niedrożnością przewodu żółciowego wspólnego.
Zmiany hematologiczne
pacjentów po rozpoczęciu leczenia fenofibratem obserwowano łagodne do umiarkowanego zmniejszenie stężenia hemoglobiny, hematokrytu i białych krwinek. Jednak poziomy te stabilizują się podczas długotrwałego podawania. U osób leczonych fenofibratem zgłaszano małopłytkowość i agranulocytozę. W ciągu pierwszych 12 miesięcy podawania produktu TRICOR zaleca się okresowe monitorowanie liczby krwinek czerwonych i białych.
Reakcje nadwrażliwości
Ostra nadwrażliwość
Po wprowadzeniu do obrotu fenofibratu zgłaszano anafilaksję i obrzęk naczynioruchowy. W niektórych przypadkach reakcje zagrażały życiu i wymagały natychmiastowego leczenia. Jeśli u pacjenta wystąpią oznaki lub objawy ostrej reakcji nadwrażliwości, należy zalecić natychmiastową pomoc lekarską i odstawienie fenofibratu.
Opóźniona nadwrażliwość
Po wprowadzeniu fenofibratu do obrotu zgłaszano ciężkie skórne działania niepożądane (SCAR), w tym zespół Stevensa-Johnsona, martwicę toksyczno-rozpływną naskórka oraz reakcje polekowe z eozynofilią i objawami ogólnoustrojowymi (DRESS). Przypadki DRESS były związane z reakcjami skórnymi (takimi jak wysypka lub złuszczające zapalenie skóry) oraz połączeniem eozynofilii, gorączki, zajęcia narządów ogólnoustrojowych (nerki, wątroba lub układ oddechowy). W przypadku podejrzenia SCAR należy odstawić fenofibrat i odpowiednio leczyć pacjentów.
Choroba żylno-zakrzepowo-zatorowa
badaniu FIELD zatorowość płucną (PE) i zakrzepicę żył głębokich (DVT) obserwowano częściej w grupie otrzymującej fenofibrat niż w grupie otrzymującej placebo. Spośród 9795 pacjentów włączonych do badania FIELD, 4900 było w grupie placebo i 4895 w grupie fenofibratu. W przypadku DVT wystąpiło 48 zdarzeń (1%) w grupie placebo i 67 (1%) w grupie fenofibratu (p = 0,074); aw przypadku PE wystąpiły 32 (0,7%) zdarzenia w grupie placebo i 53 (1%) w grupie fenofibratu (p = 0,022).
W Coronary Drug Project u większego odsetka pacjentów z grupy otrzymującej klofibrat wystąpiła zatorowość płucna lub zakrzepowe zapalenie żył, które zakończyły się zgonem lub które nie zakończyły się zgonem, w porównaniu z grupą placebo (5,2% w porównaniu z 3,3% po pięciu latach; p
Paradoksalne spadki poziomu cholesterolu HDL
Po wprowadzeniu produktu do obrotu i badaniach klinicznych pojawiły się doniesienia o ciężkim obniżeniu poziomu cholesterolu HDL (do 2 mg/dl) u pacjentów z cukrzycą i bez cukrzycy, rozpoczynających leczenie fibratami. Spadek HDL-C odzwierciedla spadek apolipoproteiny A1. Spadek ten odnotowano w ciągu 2 tygodni do lat po rozpoczęciu leczenia fibratami. Poziomy HDL-C pozostają obniżone do momentu wycofania terapii fibratami; odpowiedź na przerwanie terapii fibratami jest szybka i trwała. Kliniczne znaczenie tego spadku HDL-C nie jest znane. Zaleca się sprawdzanie poziomu HDL-C w ciągu pierwszych kilku miesięcy po rozpoczęciu terapii fibratami. W przypadku wykrycia silnie obniżonego poziomu HDL-C należy przerwać terapię fibratami i monitorować poziom HDL-C aż do powrotu do wartości wyjściowych, a terapii fibratami nie należy wznawiać.
Toksykologia niekliniczna
Karcynogeneza i mutageneza oraz upośledzenie płodności
Przeprowadzono dwa badania rakotwórczości dietetycznej na szczurach z fenofibratem. W pierwszym 24-miesięcznym badaniu szczurom Wistar podawano fenofibrat w dawce 10, 45 i 200 mg/kg/dobę, około 0,3, 1 i 6 razy większą od maksymalnej zalecanej dawki u ludzi (MRHD) wynoszącej 300 mg fenofibratu na dobę, co odpowiada 145 mg TRICOR 160 mg dziennie, w oparciu o porównania powierzchni ciała. Przy dawce 200 mg/kg/dobę (6-krotność MRHD) częstość występowania raka wątroby była znacząco zwiększona u obu płci. Statystycznie istotny wzrost raka trzustki zaobserwowano u mężczyzn przy 1 i 6 krotności MRHD; zaobserwowano wzrost liczby gruczolaków trzustki i łagodnych guzów jąder z komórek śródmiąższowych przy 6-krotnej MRHD u mężczyzn. W drugim 24-miesięcznym badaniu rakotwórczości na szczurach, przeprowadzonym na innym szczepie szczurów (Sprague-Dawley), dawki 10 i 60 mg/kg/dobę (0,3 i 2 razy większe niż MRHD) powodowały istotny wzrost częstości występowania gruczolaków groniastych trzustki u obu płci i wzrost guzów komórek śródmiąższowych jąder u mężczyzn przy 2-krotności MRHD.
Przeprowadzono 117-tygodniowe badanie rakotwórczości na szczurach, porównując trzy leki: fenofibrat 10 i 60 mg/kg/dobę (0,3 i 2 razy MRHD, na podstawie porównania powierzchni ciała), klofibrat (400 mg/kg/dobę; 2 razy dawki dla ludzi) oraz gemfibrozylu (250 mg/kg/dobę; dwukrotność dawki dla człowieka, w przeliczeniu na mg/m² powierzchni). Fenofibrat zwiększał występowanie gruczolaków groniastych trzustki u obu płci. Klofibrat powodował wzrost raka wątrobowokomórkowego i gruczolaków groniastych trzustki u mężczyzn oraz guzków nowotworowych wątroby u kobiet. Gemfibrozyl zwiększał guzki nowotworowe wątroby u mężczyzn i kobiet, podczas gdy wszystkie trzy leki zwiększały guzy komórek śródmiąższowych jąder u mężczyzn.
21-miesięcznym badaniu na myszach CF-1 fenofibrat w dawce 10, 45 i 200 mg/kg/dobę (około 0,2, 1 i 3 razy MRHD, w oparciu o porównania powierzchni ciała) znacząco zwiększał raka wątroby w obu przypadkach. płci na poziomie 3 razy MRHD. W drugim 18-miesięcznym badaniu przy dawkach 10, 60 i 200 mg/kg/dobę fenofibrat znacząco zwiększał liczbę nowotworów wątroby u samców myszy i gruczolaków wątroby u samic myszy przy trzykrotnej MRHD.
Badania mikroskopii elektronowej wykazały proliferację peroksysomów po podaniu szczurom fenofibratu. Nie przeprowadzono odpowiedniego badania w celu zbadania proliferacji peroksysomów u ludzi, ale zaobserwowano zmiany w morfologii i liczbie peroksysomów u ludzi po leczeniu innymi członkami klasy fibratów, gdy porównywano biopsje wątroby przed i po leczeniu tego samego osobnika.
Wykazano, że fenofibrat jest pozbawiony potencjału mutagennego w następujących testach: Amesa, chłoniaka myszy, aberracji chromosomowych i nieplanowanej syntezy DNA w pierwotnych hepatocytach szczura.
W badaniach płodności szczurom podawano doustnie dietetyczne dawki fenofibratu, samce otrzymywały 61 dni przed kryciem, a samice 15 dni przed kryciem poprzez odstawienie od matki, co nie powodowało niekorzystnego wpływu na płodność przy dawkach do 300 mg/kg/dobę (10-krotność MRHD, na podstawie porównań powierzchni ciała).
Używaj w określonych populacjach
Ciąża
Podsumowanie ryzyka
Ograniczone dostępne dane dotyczące stosowania fenofibratu u kobiet w ciąży są niewystarczające do określenia związanego z lekiem ryzyka poważnych wad wrodzonych, poronienia lub niekorzystnych skutków dla matki lub płodu. W badaniach na zwierzętach dotyczących reprodukcji nie zaobserwowano toksycznego działania na zarodek i płód po doustnym podaniu fenofibratu szczurom i królikom w okresie organogenezy w dawkach mniejszych lub równoważnych maksymalnej zalecanej dawce klinicznej 145 mg na dobę, w oparciu o powierzchnię ciała (mg/ m²). Niekorzystne skutki dla reprodukcji wystąpiły przy wyższych dawkach w obecności toksyczności matczynej (patrz Dane ). TRICOR 200mg powinien być stosowany w czasie ciąży tylko wtedy, gdy potencjalna korzyść uzasadnia potencjalne ryzyko dla płodu.
Szacowane ryzyko tła poważnych wad wrodzonych i poronienia dla wskazanej populacji jest nieznane. W ogólnej populacji USA szacowane podstawowe ryzyko poważnych wad wrodzonych i poronienia w klinicznie rozpoznanych ciążach wynosi odpowiednio 2-4% i 15-20%.
Dane
Dane zwierząt
ciężarnych samic szczura, którym w okresie organogenezy podawano doustnie dawki dietetyczne wynoszące 14, 127 i 361 mg/kg mc./dobę od 6-15. przy maksymalnej zalecanej dawce u ludzi [MRHD] wynoszącej 300 mg fenofibratu na dobę, co odpowiada 145 mg preparatu TRICOR na dobę, na podstawie porównań powierzchni ciała). Nasilenie wad rozwojowych szkieletu płodu obserwowano przy dawkach toksycznych dla matki (361 mg/kg/dobę, co odpowiada 12-krotnej ekspozycji klinicznej na MRHD), które znacząco hamowały przyrost masy ciała matki.
ciężarnych królików, którym w okresie organogenezy podawano doustnie przez zgłębnik dawki 15, 150 i 300 mg/kg/dobę w okresie organogenezy i którym pozwolono na poród, nie zaobserwowano niekorzystnych zmian rozwojowych przy dawce 15 mg/kg/dobę (dawka, która przybliża ekspozycję kliniczną przy MRHD, na podstawie porównań powierzchni ciała). Poronione mioty obserwowano przy dawkach toksycznych dla matek (≥ 150 mg/kg/dobę, co odpowiada ≥ 10-krotnej ekspozycji klinicznej przy MRHD), które hamowały przyrost masy ciała matki.
ciężarnych szczurów, którym podawano doustnie dawki dietetyczne 15, 75 i 300 mg/kg/dobę od 15. dnia ciąży do 21. dnia laktacji (odstawienie od piersi), nie zaobserwowano działań niepożądanych po podaniu dawki 15 mg/kg/dobę (mniej niż ekspozycja kliniczna). przy MRHD, na podstawie porównań powierzchni ciała), pomimo toksyczności matczynej (zmniejszony przyrost masy ciała). Utratę po implantacji obserwowano przy dawce ≥ 75 mg/kg/dobę (≥ 2-krotność ekspozycji klinicznej na MRHD) w obecności toksyczności matczynej (zmniejszony przyrost masy ciała). Zmniejszone przeżycie potomstwa odnotowano przy dawce 300 mg/kg/dobę (10-krotność ekspozycji klinicznej przy MRHD), co wiązało się ze zmniejszonym przyrostem masy ciała matki/zaniedbaniem matki.
Laktacja
Podsumowanie ryzyka
Brak dostępnych informacji na temat obecności fenofibratu w mleku ludzkim, wpływu leku na niemowlę karmione piersią ani wpływu na produkcję mleka. Fenofibrat jest obecny w mleku szczurów i dlatego prawdopodobnie jest obecny w mleku ludzkim. Ze względu na możliwość wystąpienia poważnych działań niepożądanych u niemowląt karmionych piersią, takich jak zaburzenia metabolizmu lipidów niemowląt, kobiety nie powinny karmić piersią podczas leczenia preparatem TRICOR 200 mg i przez 5 dni po podaniu ostatniej dawki [patrz PRZECIWWSKAZANIA ].
Zastosowanie pediatryczne
Nie ustalono bezpieczeństwa i skuteczności u pacjentów pediatrycznych.
Zastosowanie geriatryczne
Wiadomo, że kwas fenofibrynowy jest w znacznym stopniu wydalany przez nerki, a ryzyko działań niepożądanych tego leku może być większe u pacjentów z zaburzeniami czynności nerek. Wiek nie ma wpływu na ekspozycję na kwas fenofibrynowy. Ponieważ pacjenci w podeszłym wieku mają większą częstość występowania zaburzeń czynności nerek, doboru dawki u pacjentów w podeszłym wieku należy dokonywać na podstawie czynności nerek [patrz DAWKOWANIE I SPOSÓB PODAWANIA oraz FARMAKOLOGIA KLINICZNA ]. Pacjenci w podeszłym wieku z prawidłową czynnością nerek nie powinni wymagać modyfikacji dawki. Rozważ monitorowanie czynności nerek u pacjentów w podeszłym wieku przyjmujących TRICOR.
Zaburzenia czynności nerek
Należy unikać stosowania produktu TRICOR 160 mg u pacjentów z ciężkimi zaburzeniami czynności nerek [patrz PRZECIWWSKAZANIA ]. Zmniejszenie dawki jest wymagane u pacjentów z łagodnymi do umiarkowanych zaburzeniami czynności nerek [patrz DAWKOWANIE I SPOSÓB PODAWANIA oraz FARMAKOLOGIA KLINICZNA ]. Zaleca się monitorowanie czynności nerek u pacjentów z zaburzeniami czynności nerek.
Niewydolność wątroby
Stosowanie preparatu TRICOR 200 mg nie było oceniane u osób z zaburzeniami czynności wątroby [patrz PRZECIWWSKAZANIA oraz FARMAKOLOGIA KLINICZNA ].
PRZEDAWKOWAĆ
Nie ma specyficznego leczenia przedawkowania preparatu TRICOR. Wskazana jest ogólna wspomagająca opieka nad pacjentem, w tym monitorowanie parametrów życiowych i obserwacja stanu klinicznego w przypadku przedawkowania. Jeśli jest to wskazane, niewchłonięty lek należy usunąć przez wymioty lub płukanie żołądka; należy zachować zwykłe środki ostrożności w celu utrzymania dróg oddechowych. Ponieważ kwas fenofibrynowy silnie wiąże się z białkami osocza, nie należy brać pod uwagę hemodializy.
PRZECIWWSKAZANIA
TRICOR 200mg jest przeciwwskazany w:
- pacjenci z ciężkimi zaburzeniami czynności nerek, w tym pacjenci poddawani dializie [patrz FARMAKOLOGIA KLINICZNA ].
- pacjenci z czynną chorobą wątroby, w tym z pierwotną marskością żółciową wątroby i niewyjaśnionymi uporczywymi zaburzeniami czynności wątroby [patrz OSTRZEŻENIA I ŚRODKI ].
- pacjenci z istniejącą wcześniej chorobą pęcherzyka żółciowego [patrz OSTRZEŻENIA I ŚRODKI ].
- karmiące matki [patrz Używaj w określonych populacjach ].
- pacjenci ze stwierdzoną nadwrażliwością na fenofibrat lub kwas fenofibrynowy [patrz OSTRZEŻENIA I ŚRODKI ].
FARMAKOLOGIA KLINICZNA
Mechanizm akcji
Aktywną częścią TRICOR jest kwas fenofibrynowy. Farmakologiczne działanie kwasu fenofibrynowego zarówno u zwierząt, jak iu ludzi było szeroko badane po podaniu doustnym fenofibratu.
Obserwowane w praktyce klinicznej działanie modyfikujące lipidy kwasu fenofibrynowego zostało wyjaśnione in vivo u myszy transgenicznych oraz in vitro w hodowlach ludzkich hepatocytów poprzez aktywację receptora a aktywowanego przez proliferatory peroksysomów (PPARα). Dzięki temu mechanizmowi fenofibrat zwiększa lipolizę i eliminację cząstek bogatych w triglicerydy z osocza poprzez aktywację lipazy lipoproteinowej i zmniejszenie wytwarzania apoproteiny C-III (inhibitor aktywności lipazy lipoproteinowej).
Wynikający z tego spadek TG powoduje zmianę wielkości i składu LDL z małych, gęstych cząstek (które uważa się za miażdżycogenne ze względu na ich podatność na utlenianie) do dużych cząstek unoszących się na powierzchni. Te większe cząstki mają większe powinowactwo do receptorów cholesterolu i są szybko katabolizowane. Aktywacja PPARα powoduje również wzrost syntezy apolipoprotein AI, A-II i HDL-cholesterolu.
Fenofibrat zmniejsza również stężenie kwasu moczowego w surowicy u osób z hiperurycemią i zdrowych, zwiększając wydalanie kwasu moczowego z moczem.
Farmakodynamika
Różne badania kliniczne wykazały, że podwyższone poziomy całkowitego C, LDL-C i apo B, kompleksu błonowego LDL, są związane z miażdżycą u ludzi. Podobnie, obniżone poziomy HDL-C i jego kompleksu transportowego, apolipoproteiny A (apo AI i apo AII) są związane z rozwojem miażdżycy. Badania epidemiologiczne wykazały, że chorobowość i śmiertelność z przyczyn sercowo-naczyniowych zależy bezpośrednio od poziomu C całkowitego, LDL-C i TG oraz odwrotnie z poziomem HDL-C. Nie określono niezależnego wpływu podwyższenia HDL-C lub obniżenia stężenia triglicerydów (TG) na ryzyko zachorowalności i śmiertelności z przyczyn sercowo-naczyniowych.
Kwas fenofibrynowy, aktywny metabolit fenofibratu, powoduje zmniejszenie stężenia cholesterolu całkowitego, cholesterolu LDL, apolipoproteiny B, triglicerydów całkowitych i lipoproteiny bogatej w triglicerydy (VLDL) u leczonych pacjentów. Ponadto leczenie fenofibratem powoduje wzrost lipoprotein o wysokiej gęstości (HDL) oraz apolipoprotein apoAI i apoAII.
Farmakokinetyka
Stężenia kwasu fenofibrynowego w osoczu po podaniu trzech tabletek 48 mg lub jednej tabletki 145 mg są równoważne w warunkach po posiłku jednej 200 mg mikronizowanej kapsułce fenofibratu.
Fenofibrat jest prolekiem aktywnej cząsteczki chemicznej kwasu fenofibrynowego. Fenofibrat jest przekształcany w organizmie na drodze hydrolizy estrów do kwasu fenofibrynowego, który jest aktywnym składnikiem mierzalnym w krążeniu.
Wchłanianie
Nie można określić bezwzględnej biodostępności fenofibratu, ponieważ związek jest praktycznie nierozpuszczalny w środowisku wodnym odpowiednim do wstrzykiwania. Jednak fenofibrat jest dobrze wchłaniany z przewodu pokarmowego. Po podaniu doustnym zdrowym ochotnikom około 60% pojedynczej dawki znakowanego radioizotopem fenofibratu pojawiło się w moczu, głównie w postaci kwasu fenofibrynowego i jego koniugatu glukuronianowego, a 25% zostało wydalone z kałem. Maksymalne stężenie kwasu fenofibrynowego w osoczu występuje w ciągu 6 do 8 godzin po podaniu.
Ekspozycja na kwas fenofibrynowy w osoczu, mierzona jako Cmax i AUC, nie różni się znacząco w przypadku podania pojedynczej dawki 145 mg fenofibratu na czczo lub nie na czczo.
Dystrybucja
Po wielokrotnym podaniu fenofibratu stan stacjonarny kwasu fenofibrynowego osiągany jest w ciągu 9 dni. Stężenia kwasu fenofibrynowego w osoczu w stanie stacjonarnym są w przybliżeniu dwukrotnie wyższe niż po podaniu pojedynczej dawki. Wiązanie z białkami surowicy wynosiło około 99% u osób zdrowych i z hiperlipidemią.
Metabolizm
Po podaniu doustnym fenofibrat jest szybko hydrolizowany przez esterazy do aktywnego metabolitu, kwasu fenofibrynowego; niezmieniony fenofibrat nie jest wykrywany w osoczu.
Kwas fenofibrynowy jest głównie sprzężony z kwasem glukuronowym, a następnie wydalany z moczem. Niewielka ilość kwasu fenofibrynowego jest redukowana w grupie karbonylowej do metabolitu benzhydrolowego, który z kolei jest skoniugowany z kwasem glukuronowym i wydalany z moczem.
Dane dotyczące metabolizmu in vivo wskazują, że ani fenofibrat, ani kwas fenofibrynowy nie ulegają metabolizmowi oksydacyjnemu (np. cytochrom P450) w istotnym stopniu.
Eliminacja
Po wchłonięciu fenofibrat jest wydalany głównie z moczem w postaci metabolitów, głównie kwasu fenofibrynowego i glukuronidu kwasu fenofibrynowego. Po podaniu znakowanego radioizotopem fenofibratu około 60% dawki pojawiło się w moczu, a 25% zostało wydalone z kałem.
Kwas fenofibrynowy jest eliminowany z okresem półtrwania wynoszącym 20 godzin, co pozwala na dawkowanie raz na dobę.
Specjalne populacje
Geriatria
U ochotników w podeszłym wieku w wieku od 77 do 87 lat klirens kwasu fenofibrynowego po podaniu pojedynczej dawki doustnej fenofibratu wynosił 1,2 l/h, w porównaniu do 1,1 l/h u młodych dorosłych. Wskazuje to, że podobny schemat dawkowania można stosować u osób starszych z prawidłową czynnością nerek, bez zwiększania akumulacji leku lub metabolitów [patrz DAWKOWANIE I SPOSÓB PODAWANIA oraz Używaj w określonych populacjach ].
Pediatria
Nie badano farmakokinetyki produktu TRICOR 160 mg w populacjach pediatrycznych.
Płeć
Nie zaobserwowano różnic w farmakokinetyce fenofibratu między mężczyznami i kobietami.
Wyścig
Nie badano wpływu rasy na farmakokinetykę fenofibratu, jednak fenofibrat nie jest metabolizowany przez enzymy, o których wiadomo, że wykazują zmienność międzyetniczną.
Zaburzenia czynności nerek
Farmakokinetykę kwasu fenofibrynowego badano u pacjentów z łagodnymi, umiarkowanymi i ciężkimi zaburzeniami czynności nerek. U pacjentów z ciężkimi zaburzeniami czynności nerek (szacowany współczynnik przesączania kłębuszkowego [eGFR] DAWKOWANIE I SPOSÓB PODAWANIA ].
Niewydolność wątroby
Nie przeprowadzono badań farmakokinetycznych u pacjentów z zaburzeniami czynności wątroby.
Interakcje lekowe
Badania in vitro z użyciem mikrosomów wątroby ludzkiej wskazują, że fenofibrat i kwas fenofibrynowy nie są inhibitorami izoform cytochromu (CYP) P450 CYP3A4, CYP2D6, CYP2E1 ani CYP1A2. Są słabymi inhibitorami CYP2C8, CYP2C19 i CYP2A6 oraz łagodnymi do umiarkowanych inhibitorami CYP2C9 w stężeniach terapeutycznych.
Tabela 2 opisuje wpływ leków podawanych jednocześnie na ogólnoustrojową ekspozycję na kwas fenofibrynowy. Tabela 3 opisuje wpływ jednoczesnego podawania fenofibratu lub kwasu fenofibrynowego na inne leki.
Studia kliniczne
Pierwotna hipercholesterolemia (heterozygotyczna rodzinna i nierodzinna) oraz mieszana dyslipidemia
Działanie fenofibratu w dawce odpowiadającej 145 mg TRICOR (tabletki fenofibratu) na dobę oceniano w czterech randomizowanych, kontrolowanych placebo badaniach z podwójnie ślepą próbą, prowadzonych w grupach równoległych, obejmujących pacjentów z następującymi średnimi wyjściowymi wartościami lipidów: C 306,9 całkowite mg/dl; LDL-C 213,8 mg/dl; HDL-C 52,3 mg/dl; i triglicerydy 191,0 mg/dl. Terapia TRICOR 200 mg obniżyła LDL-C, Całkowity C i stosunek LDL-C/HDL-C. Terapia TRICOR również obniżyła triglicerydy i podwyższyła HDL-C (patrz Tabela 4).
W podgrupie badanych przeprowadzono pomiary apo B. Leczenie preparatem TRICOR 200 mg istotnie zmniejszyło apo B od wartości początkowej do punktu końcowego w porównaniu z placebo (-25,1% w porównaniu z 2,4%, p
Ciężka hipertriglicerydemia
Wpływ fenofibratu na triglicerydy w surowicy badano w dwóch randomizowanych, podwójnie zaślepionych, kontrolowanych placebo badaniach klinicznych z udziałem 147 pacjentów z hipertriglicerydemią. Pacjenci byli leczeni przez osiem tygodni według protokołów, które różniły się tylko tym, że jeden przyjmował pacjentów z wyjściowymi poziomami TG od 500 do 1500 mg/dl, a drugimi z poziomami TG od 350 do 500 mg/dl. U pacjentów z hipertriglicerydemią i prawidłową cholesterolemią z hiperchylomikronemią lub bez niej leczenie fenofibratem w dawkach równoważnych TRICOR 145 mg na dobę zmniejszało głównie stężenie triglicerydów lipoprotein o bardzo małej gęstości (VLDL) i cholesterolu VLDL. Leczenie pacjentów z podwyższonym stężeniem triglicerydów często prowadzi do wzrostu LDL-C (patrz Tabela 5).
Wpływ TRICOR 160mg na zachorowalność i śmiertelność sercowo-naczyniową nie został określony.
INFORMACJA O PACJENCIE
Pacjentów należy poinformować:
- potencjalnych korzyści i zagrożeń związanych z TRICOR.
- nie stosować leku TRICOR w przypadku znanej nadwrażliwości na fenofibrat lub kwas fenofibrynowy.
- leków, które nie powinny być przyjmowane w połączeniu z TRICOR.
- że jeśli przyjmują antykoagulanty kumaryny, TRICOR 160 mg może nasilać ich działanie przeciwzakrzepowe i może być konieczne wzmożone monitorowanie.
- aby kontynuować przestrzeganie odpowiedniej diety modyfikującej lipidy podczas przyjmowania leku TRICOR.
- przyjmować TRICOR 160 mg raz na dobę, niezależnie od posiłków, w przepisanej dawce, połykając każdą tabletkę w całości.
- wrócić do gabinetu lekarskiego w celu rutynowego monitorowania.
- informować swojego lekarza o wszystkich przyjmowanych lekach, suplementach i preparatach ziołowych oraz o wszelkich zmianach ich stanu zdrowia. Pacjentom należy również doradzić, aby poinformowali swoich lekarzy przepisujących nowy lek, że przyjmują TRICOR.
- poinformować lekarza o objawach uszkodzenia wątroby (np. żółtaczka, ból brzucha, nudności, złe samopoczucie, ciemny mocz, nieprawidłowy stolec, świąd); jakikolwiek ból mięśni, tkliwość lub osłabienie; lub jakiekolwiek inne nowe objawy.
- nie karmić piersią podczas leczenia lekiem TRICOR i przez 5 dni po przyjęciu ostatniej dawki.