Xeloda 500mg Capecitabine Zastosowanie, skutki uboczne i dawkowanie. Cena w aptece internetowej. Leki generyczne bez recepty.
Co to jest Xeloda 500mg i jak się go stosuje?
Xeloda to lek na receptę stosowany w leczeniu objawów nowotworów, takich jak rak okrężnicy, rak jelita grubego i rak piersi. Xeloda można stosować samodzielnie lub z innymi lekami.
Xeloda należy do klasy leków o nazwie Przeciwnowotworowe, Antymetabolity.
Nie wiadomo, czy Xeloda 500mg jest bezpieczna i skuteczna u dzieci.
Jakie są możliwe skutki uboczne Xeloda 500mg?
Xeloda może powodować poważne działania niepożądane, w tym:
- gorączka powyżej 100,5 stopnia,
- mdłości,
- utrata apetytu,
- jeść znacznie mniej niż zwykle,
- wymioty (więcej niż raz na 24 godziny),
- ciężka biegunka (więcej niż 4 razy dziennie lub w nocy),
- pęcherze lub owrzodzenia w jamie ustnej,
- zaczerwienione lub opuchnięte dziąsła,
- kłopoty z połykaniem,
- ból, tkliwość, zaczerwienienie, obrzęk, pęcherze lub łuszczenie się skóry dłoni lub stóp,
- uczucie pragnienia lub gorąca,
- niemożność oddania moczu,
- silne pocenie się,
- gorąca i sucha skóra,
- ból lub ucisk w klatce piersiowej,
- nierówne bicie serca,
- duszność,
- obrzęk lub szybki przyrost masy ciała,
- bolesne lub utrudnione oddawanie moczu,
- obrzęk stóp lub kostek,
- czuć się zmęczonym,
- duszność,
- ciemny mocz,
- stołki w kolorze gliny,
- zażółcenie skóry lub oczu (żółtaczka),
- gorączka lub inne objawy grypy,
- kaszel,
- owrzodzenia skóry,
- blada skóra,
- łatwe siniaki,
- nietypowe krwawienie,
- uczucie oszołomienia,
- szybkie tętno,
- ból gardła,
- obrzęk twarzy lub języka,
- pieczenie w twoich oczach i
- ból skóry, po którym następuje czerwona lub fioletowa wysypka (zwłaszcza na twarzy lub górnej części ciała) i powoduje powstawanie pęcherzy i złuszczanie
Natychmiast uzyskaj pomoc medyczną, jeśli wystąpi którykolwiek z wymienionych powyżej objawów.
Najczęstsze skutki uboczne Xeloda 500mg to:
- ból brzucha,
- zaparcie,
- niestrawność,
- uczucie zmęczenia,
- łagodna wysypka skórna i
- drętwienie lub mrowienie w dłoniach lub stopach
Poinformuj lekarza, jeśli wystąpią jakiekolwiek skutki uboczne, które Ci przeszkadzają lub które nie ustępują.
To nie wszystkie możliwe skutki uboczne Xeloda. Aby uzyskać więcej informacji, skontaktuj się z lekarzem lub farmaceutą.
Zadzwoń do lekarza, aby uzyskać poradę medyczną na temat skutków ubocznych. Możesz zgłosić skutki uboczne do FDA pod numerem 1-800-FDA-1088.
OSTRZEŻENIE
INTERAKCJA XELODA-WARFARYNA
XELODA 500 mg Warfaryna Interakcja: U pacjentów otrzymujących jednocześnie kapecytabinę i doustne leki przeciwzakrzepowe pochodne kumaryny należy często monitorować odpowiedź przeciwzakrzepową (INR lub czas protrombinowy) w celu odpowiedniego dostosowania dawki leku przeciwzakrzepowego. Klinicznie istotna interakcja leku XELODA-warfaryna została wykazana w badaniu farmakologii klinicznej [patrz OSTRZEŻENIA I ŚRODKI INTERAKCJE Z LEKAMI]. U pacjentów przyjmujących XELODA 500 mg jednocześnie z lekami przeciwzakrzepowymi będącymi pochodnymi kumaryny, takimi jak warfaryna i fenprokumon, zgłaszano zmiany parametrów krzepnięcia i (lub) krwawienia, w tym zgon. Raporty po wprowadzeniu produktu do obrotu wykazały klinicznie istotne wydłużenie czasu protrombinowego (PT) i INR u pacjentów, u których w momencie wprowadzenia produktu XELODA 500 mg ustabilizowano leki przeciwzakrzepowe. Zdarzenia te wystąpiły w ciągu kilku dni i do kilku miesięcy po rozpoczęciu leczenia XELODA 500 mg, a w kilku przypadkach w ciągu 1 miesiąca po zaprzestaniu leczenia XELODA. Zdarzenia te wystąpiły u pacjentów z przerzutami do wątroby i bez przerzutów. Wiek powyżej 60 lat i rozpoznanie raka niezależnie predysponują pacjentów do zwiększonego ryzyka koagulopatii.
OPIS
XELODA (kapecytabina) to karbaminian fluoropirymidyny o działaniu przeciwnowotworowym. Jest to podawany doustnie ogólnoustrojowy prolek 5'-deoksy-5-fluorourydyny (5'-DFUR), który jest przekształcany w 5-fluorouracyl.
Nazwa chemiczna kapecytabiny to 5'-deoksy-5-fluoro-N-[(pentyloksy)karbonylo]-cytydyna i ma masę cząsteczkową 359,35. Kapecytabina ma następujący wzór strukturalny:
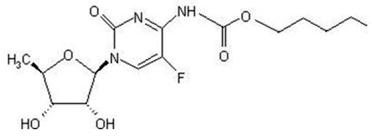
Kapecytabina jest białym lub prawie białym krystalicznym proszkiem o rozpuszczalności w wodzie 26 mg/ml w temperaturze 20°C.
XELODA jest dostarczany w postaci dwuwypukłych, podłużnych tabletek powlekanych do podawania doustnego. Każda jasna tabletka koloru brzoskwiniowego zawiera 150 mg kapecytabiny, a każda tabletka koloru brzoskwiniowego zawiera 500 mg kapecytabiny. Nieaktywne składniki preparatu XELODA to: laktoza bezwodna, kroskarmeloza sodowa, hydroksypropylometyloceluloza, celuloza mikrokrystaliczna, stearynian magnezu i woda oczyszczona. Powłoka brzoskwiniowa lub jasna brzoskwinia zawiera hydroksypropylometylocelulozę, talk, dwutlenek tytanu oraz syntetyczny żółty i czerwony tlenki żelaza.
WSKAZANIA
Rak jelita grubego
- XELODA jest wskazany jako pojedynczy środek w leczeniu uzupełniającym u pacjentów z rakiem okrężnicy typu Dukes C, którzy przeszli całkowitą resekcję guza pierwotnego, gdy preferowane jest leczenie samą fluoropirymidyną. XELODA 500 mg nie był gorszy od 5fluorouracylu i leukoworyny (5-FU/LV) pod względem przeżycia wolnego od choroby (DFS). Lekarze powinni wziąć pod uwagę wyniki badań nad chemioterapią skojarzoną, które wykazały poprawę w zakresie DFS i OS, przepisując pojedynczy środek XELODA 500 mg w leczeniu uzupełniającym raka okrężnicy typu C Dukesa.
- XELODA 500 mg jest wskazana jako leczenie pierwszego rzutu u pacjentów z rakiem jelita grubego z przerzutami, gdy preferowane jest leczenie samą fluoropirymidyną. Chemioterapia skojarzona wykazała poprawę przeżycia w porównaniu z samą 5-FU/LV. W przypadku monoterapii produktem XELODA 500 mg nie wykazano korzyści w zakresie przeżycia w porównaniu z 5-FU/LV. Stosowanie XELODA zamiast 5FU/LV w kombinacjach nie zostało odpowiednio zbadane, aby zapewnić bezpieczeństwo lub zachowanie korzyści w zakresie przeżycia.
Rak piersi
- XELODA w skojarzeniu z docetakselem jest wskazany w leczeniu pacjentów z rakiem piersi z przerzutami po niepowodzeniu wcześniejszej chemioterapii zawierającej antracykliny.
- XELODA 500 mg w monoterapii jest również wskazana w leczeniu pacjentów z rakiem piersi z przerzutami, opornym zarówno na paklitaksel, jak i na schemat chemioterapii zawierającym antracykliny lub opornym na paklitaksel i u których dalsze leczenie antracyklinami nie jest wskazane (np. pacjentki, które otrzymały skumulowane dawki 400 mg/m2 doksorubicyny lub ekwiwalentów doksorubicyny). Oporność definiuje się jako postępującą chorobę w trakcie leczenia, z początkową odpowiedzią lub bez, lub nawrót w ciągu 6 miesięcy od zakończenia leczenia adiuwantowym schematem zawierającym antracykliny.
DAWKOWANIE I SPOSÓB PODAWANIA
Ważne instrukcje administracyjne
Tabletki XELODA należy połykać w całości popijając wodą w ciągu 30 minut po posiłku. XELODA 500mg jest lekiem cytotoksycznym. Należy postępować zgodnie z odpowiednimi specjalnymi procedurami postępowania i usuwania.1 Jeśli konieczne jest przecięcie lub zgniecenie tabletek XELODA 500 mg, powinien to zrobić profesjonalista przeszkolony w zakresie bezpiecznego obchodzenia się z lekami cytotoksycznymi przy użyciu odpowiedniego sprzętu i procedur bezpieczeństwa. Dawkę produktu XELODA oblicza się na podstawie powierzchni ciała.
Standardowa dawka początkowa
Monoterapia (przerzutowy rak jelita grubego, adjuwantowy rak jelita grubego, rak piersi z przerzutami)
Zalecana dawka produktu XELODA to 1250 mg/m2 podawana doustnie dwa razy na dobę (rano i wieczorem; co odpowiada całkowitej dawce dobowej 2500 mg/m2) przez 2 tygodnie, po których następuje 1-tygodniowy okres odpoczynku podawany jako 3-tygodniowe cykle (patrz ).
Leczenie uzupełniające u pacjentów z rakiem okrężnicy typu Dukes C jest zalecane przez łącznie 6 miesięcy [tj. XELODA 1250 mg/m2 doustnie dwa razy dziennie przez 2 tygodnie, po czym następuje 1-tygodniowy okres odpoczynku, podawany jako 3-tygodniowe cykle, łącznie 8 cykli (24 tygodnie)].
W połączeniu z docetakselem (rak piersi z przerzutami)
W skojarzeniu z docetakselem zalecana dawka produktu XELODA wynosi 1250 mg/m2 dwa razy na dobę przez 2 tygodnie, po czym następuje 1 tydzień przerwy, w połączeniu z docetakselem w dawce 75 mg/m2 w postaci 1-godzinnego wlewu dożylnego co 3 tygodnie. Premedykację, zgodnie z oznakowaniem docetakselu, należy rozpocząć przed podaniem docetakselu u pacjentów otrzymujących produkt XELODA w połączeniu z docetakselem. wyświetla całkowitą dawkę dobową produktu XELODA według powierzchni ciała oraz liczbę tabletek, które należy przyjąć przy każdej dawce.
Wytyczne dotyczące zarządzania dawką
Ogólny
Dawkowanie produktu XELODA może wymagać indywidualnego dostosowania, aby zoptymalizować postępowanie z pacjentem. Pacjentów należy uważnie monitorować pod kątem toksyczności, a dawki produktu XELODA 500 mg należy w razie potrzeby modyfikować w celu dostosowania indywidualnej tolerancji pacjenta na leczenie [patrz Studia kliniczne ]. Toksyczność spowodowaną podaniem produktu XELODA 500 mg można opanować poprzez leczenie objawowe, przerwanie dawkowania i dostosowanie dawki produktu XELODA. Po zmniejszeniu dawki nie należy jej zwiększać w późniejszym czasie. Dawki XELODA 500mg pominięte ze względu na toksyczność nie są zastępowane ani przywracane; zamiast tego pacjent powinien wznowić zaplanowane cykle leczenia.
Może zaistnieć potrzeba zmniejszenia dawki fenytoiny i leków przeciwzakrzepowych będących pochodnymi kumaryny, jeśli którykolwiek z leków podawany jest jednocześnie z produktem XELODA [patrz INTERAKCJE Z LEKAMI ].
Monoterapia (przerzutowy rak jelita grubego, adjuwantowy rak jelita grubego, rak piersi z przerzutami)
Schemat modyfikacji dawki produktu XELODA opisany poniżej (patrz ) jest zalecany do postępowania w przypadku działań niepożądanych.
W połączeniu z docetakselem (rak piersi z przerzutami)
Modyfikacje dawki preparatu XELODA 500mg ze względu na toksyczność należy wykonać zgodnie z powyższym dla preparatu XELODA. Na początku cyklu leczenia, jeśli wskazane jest opóźnienie leczenia albo XELODA 500 mg, albo docetakselem, podanie obu leków należy opóźnić do czasu spełnienia wymagań ponownego uruchomienia obu leków.
Schemat zmniejszania dawki docetakselu stosowanego w skojarzeniu z produktem XELODA w leczeniu przerzutowego raka piersi przedstawiono w Tabeli 3.
Dostosowanie dawki początkowej w specjalnych populacjach
Zaburzenia czynności nerek
Nie zaleca się dostosowywania dawki początkowej produktu XELODA 500 mg u pacjentów z łagodnymi zaburzeniami czynności nerek (klirens kreatyniny = 51 do 80 ml/min [Cockroft i Gault, jak pokazano poniżej]). U pacjentów z umiarkowanymi zaburzeniami czynności nerek (wyjściowy klirens kreatyniny = 30 do 50 ml/min) zmniejszenie dawki do 75% dawki początkowej produktu XELODA w przypadku stosowania w monoterapii lub w skojarzeniu z docetakselem (z 1250 mg/m2 do 950 mg/m2 dwa razy dziennie) jest zalecane [patrz Używaj w określonych populacjach oraz FARMAKOLOGIA KLINICZNA ]. Zaleca się późniejszą modyfikację dawki zgodnie z opisem i (w zależności od schematu), jeśli u pacjenta wystąpi zdarzenie niepożądane stopnia 2 do 4 [patrz OSTRZEŻENIA I ŚRODKI ]. Zalecenia dotyczące dostosowania dawki początkowej u pacjentów z umiarkowanymi zaburzeniami czynności nerek dotyczą zarówno produktu XELODA 500 mg w monoterapii, jak i produktu XELODA 500 mg w skojarzeniu z docetakselem.
Równanie Cockrofta i Gaulta:
Geriatria
Lekarze powinni zachować ostrożność podczas monitorowania działania leku XELODA 500 mg u osób w podeszłym wieku. Dostępne są niewystarczające dane, aby podać zalecenia dotyczące dawkowania.
JAK DOSTARCZONE
Formy dawkowania Mocne strony
XELODA jest dostarczany w postaci dwuwypukłych, podłużnych tabletek powlekanych do podawania doustnego. Każda tabletka koloru jasnej brzoskwini zawiera 150 mg kapecytabiny, a każda tabletka koloru brzoskwini zawiera 500 mg kapecytabiny.
150 mg
Barwa: Jasnobrzoskwiniowa Grawerowanie: XELODA z jednej strony i 150 z drugiej 150 mg tabletki pakowane są w butelki po 60 ( NDC 0004-1100-20), pakowane pojedynczo w karton.
500 mg
Kolor: brzoskwiniowy Grawerowanie: XELODA 500mg z jednej strony i 500mg z drugiej 500mg tabletki pakowane są w butelki po 120 ( NDC 0004-1101-50), pakowane pojedynczo w karton.
Składowania i stosowania
Przechowywać w 25°C (77°F); wycieczki dozwolone do 15 ° do 30 ° C (59 ° do 86 ° F). [Patrz Kontrolowana temperatura pomieszczenia USP]. TRZYMAĆ SZCZELNIE ZAMKNIĘTE.
XELODA to lek cytotoksyczny. Należy postępować zgodnie z odpowiednimi specjalnymi procedurami postępowania i utylizacji.1 Wszelkie niewykorzystane resztki produktu należy usunąć zgodnie z lokalnymi przepisami lub zgodnie z programami zwrotu leków.
BIBLIOGRAFIA
1. „Niebezpieczne leki OSHA”. OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
Dystrybutor: Genentech USA, Inc. Członek grupy Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990. Poprawiono: maj 2021
SKUTKI UBOCZNE
Ponieważ badania kliniczne prowadzone są w bardzo zróżnicowanych warunkach, częstość występowania działań niepożądanych obserwowanych w badaniach klinicznych leku nie może być bezpośrednio porównywana z częstością w badaniach klinicznych innego leku i może nie odzwierciedlać częstości obserwowanych w praktyce.
Adiuwantowy rak okrężnicy
przedstawia działania niepożądane występujące u ≥5% pacjentów z jednego badania fazy 3 u pacjentów z rakiem okrężnicy typu Dukes C, którzy otrzymali co najmniej jedną dawkę badanego leku i przeszli co najmniej jedną ocenę bezpieczeństwa. Łącznie 995 pacjentów otrzymywało 1250 mg/m2 dwa razy dziennie XELODA 500 mg podawanej przez 2 tygodnie, a następnie 1 tydzień przerwy, a 974 pacjentów otrzymywało 5-FU i leukoworynę (20 mg/m2 leukoworyny IV, a następnie 425 mg/m2 dożylnie bolus 5FU w dniach 1-5 co 28 dni). Mediana czasu trwania leczenia wyniosła 164 dni dla pacjentów leczonych kapecytabiną i 145 dni dla pacjentów leczonych 5-FU/LV. Łącznie 112 (11%) i 73 (7%) pacjentów leczonych kapecytabiną i 5-FU/LV przerwało leczenie z powodu działań niepożądanych. Łącznie 18 zgonów ze wszystkich przyczyn wystąpiło w trakcie badania lub w ciągu 28 dni od otrzymania badanego leku: 8 (0,8%) pacjentów przydzielonych losowo do grupy XELODA 500 mg i 10 (1,0%) przydzielonych losowo do grupy 5-FU/LV.
pokazuje nieprawidłowości laboratoryjne stopnia 3/4 występujące u ≥1% pacjentów z jednego badania fazy 3 u pacjentów z rakiem okrężnicy typu Dukes C, którzy otrzymali co najmniej jedną dawkę badanego leku i mieli co najmniej jedną ocenę bezpieczeństwa.
Przerzutowy rak jelita grubego
Monoterapia
przedstawia działania niepożądane występujące u ≥5% pacjentów z połączonych dwóch badań fazy 3 w raku jelita grubego z przerzutami pierwszego rzutu. Łącznie 596 pacjentów z przerzutowym rakiem jelita grubego było leczonych 1250 mg/m2 dwa razy na dobę XELODA podawanej przez 2 tygodnie, a następnie 1-tygodniowej przerwy, a 593 pacjentom podawano 5-FU i leukoworynę w schemacie Mayo (20 dożylnie w mg/m2 leukoworyny, a następnie 425 mg/m2 dożylnie w bolusie 5-FU, w dniach 1-5, co 28 dni). W połączonej bazie danych jelita grubego mediana czasu trwania leczenia wyniosła 139 dni dla pacjentów leczonych kapecytabiną i 140 dni dla pacjentów leczonych 5-FU/LV. Łącznie 78 (13%) i 63 (11%) pacjentów leczonych kapecytabiną i 5-FU/LV przerwało leczenie z powodu działań niepożądanych lub współistniejących chorób. Łącznie 82 zgony ze wszystkich przyczyn wystąpiły w trakcie badania lub w ciągu 28 dni od otrzymania badanego leku: 50 (8,4%) pacjentów przydzielonych losowo do grupy XELODA 500 mg i 32 (5,4%) przydzielonych losowo do grupy 5-FU/LV.
Rak piersi
W połączeniu z Docetakselem
Poniższe dane przedstawiono dla badania skojarzonego produktu XELODA 500 mg i docetakselu u pacjentek z rakiem piersi z przerzutami w i . W ramieniu leczenia skojarzonego XELODA i docetaksel leczenie polegało na podawaniu produktu XELODA doustnie w dawce 1250 mg/m2 dwa razy na dobę jako terapii przerywanej (2 tygodnie leczenia, a następnie 1 tydzień bez leczenia) przez co najmniej 6 tygodni oraz docetaksel podawany w postaci 1-godzinnego wlewu dożylnego o godz. dawka 75 mg/m2 pierwszego dnia każdego 3-tygodniowego cyklu przez co najmniej 6 tygodni. W ramieniu monoterapii docetaksel podawano w 1-godzinnym wlewie dożylnym w dawce 100 mg/m2 pierwszego dnia każdego 3-tygodniowego cyklu przez co najmniej 6 tygodni. Średni czas trwania leczenia wynosił 129 dni w ramieniu leczenia skojarzonego i 98 dni w ramieniu monoterapii. Łącznie 66 pacjentów (26%) w ramieniu leczenia skojarzonego i 49 (19%) w ramieniu monoterapii wycofało się z badania z powodu działań niepożądanych. Odsetek pacjentów wymagających zmniejszenia dawki z powodu działań niepożądanych wynosił 65% w ramieniu terapii skojarzonej i 36% w ramieniu monoterapii. Odsetek pacjentów wymagających przerwania leczenia z powodu działań niepożądanych w ramieniu leczenia skojarzonego wyniósł 79%. Przerwy w leczeniu stanowiły część schematu modyfikacji dawki w grupie terapii skojarzonej, ale nie w przypadku pacjentów leczonych docetakselem w monoterapii.
Monoterapia
Poniższe dane przedstawiono dla badania u pacjentek z rakiem piersi w IV stopniu zaawansowania, które otrzymywały dawkę 1250 mg/m2 podawana dwa razy dziennie przez 2 tygodnie, po czym następowała 1-tygodniowa przerwa. Średni czas leczenia wynosił 114 dni. Łącznie 13 ze 162 pacjentów (8%) przerwało leczenie z powodu działań niepożądanych/współistniejących chorób.
Klinicznie istotne zdarzenia niepożądane u
Poniżej przedstawiono klinicznie istotne zdarzenia niepożądane zgłoszone u
Monoterapia (przerzutowy rak jelita grubego, adjuwantowy rak jelita grubego, rak piersi z przerzutami)
Przewód pokarmowy: wzdęcie brzucha, dysfagia, ból odbytu, wodobrzusze (0,1%), wrzód żołądka (0,1%), niedrożność jelit (0,3%), toksyczne rozszerzenie jelit, zapalenie żołądka i jelit (0,1%)
Skóra i skóra podskórna: zaburzenia paznokci (0,1%), zwiększone pocenie się (0,1%), nadwrażliwość na światło (0,1%), owrzodzenie skóry, świąd, zespół przypominający popromienny (0,2%)
Ogólny: ból w klatce piersiowej (0,2%), choroba grypopodobna, uderzenia gorąca, ból (0,1%), chrypka, drażliwość, trudności w chodzeniu, pragnienie, masa w klatce piersiowej, zapaść, zwłóknienie (0,1%), krwotok, obrzęk, uspokojenie
Neurologiczna: bezsenność, ataksja (0,5%), drżenie, dysfazja, encefalopatia (0,1%), nieprawidłowa koordynacja, dyzartria, utrata przytomności (0,2%), zaburzenia równowagi
Metabolizm: zwiększenie masy ciała, kacheksja (0,4%), hipertriglicerydemia (0,1%), hipokaliemia, hipomagnezemia
Oko: zapalenie spojówek
Oddechowy: kaszel (0,1%), krwawienie z nosa (0,1%), astma (0,2%), krwioplucie, niewydolność oddechowa (0,1%), duszność
Sercowy: częstoskurcz (0,1%), bradykardia, migotanie przedsionków, skurcze dodatkowe komorowe, skurcze dodatkowe, zapalenie mięśnia sercowego (0,1%), wysięk osierdziowy
Infekcje: zapalenie krtani (1,0%), zapalenie oskrzeli (0,2%), zapalenie płuc (0,2%), odoskrzelowe zapalenie płuc (0,2%), zapalenie rogówki i spojówek, posocznica (0,3%), zakażenia grzybicze (w tym kandydoza) (0,2%)
Układ mięśniowo-szkieletowy: ból mięśni, ból kości (0,1%), zapalenie stawów (0,1%), osłabienie mięśni
Krew i limfatyczny: leukopenia (0,2%), zaburzenia krzepnięcia (0,1%), zahamowanie czynności szpiku (0,1%), samoistna plamica małopłytkowa (1,0%), pancytopenia (0,1%)
Naczyniowy: niedociśnienie (0,2%), nadciśnienie (0,1%), obrzęk limfatyczny (0,1%), zator płucny (0,2%), udar naczyniowy mózgu (0,1%)
Psychiatryczny: depresja, splątanie (0,1%)
Nerkowy: zaburzenia czynności nerek (0,6%)
Ucho: zawrót głowy
Wątroby i dróg żółciowych: zwłóknienie wątroby (0,1%), zapalenie wątroby (0,1%), cholestatyczne zapalenie wątroby (0,1%), nieprawidłowe wyniki testów czynności wątroby
Układ odpornościowy: nadwrażliwość na lek (0,1%)
XELODA 500 mg w połączeniu z docetakselem (przerzutowy rak piersi)
Przewód pokarmowy: niedrożność jelit (0,4%), martwicze zapalenie jelit (0,4%), wrzód przełyku (0,4%), biegunka krwotoczna (0,8%)
Neurologiczna: ataksja (0,4%), omdlenia (1,2%), utrata smaku (0,8%), polineuropatia (0,4%), migrena (0,4%)
Sercowy: częstoskurcz nadkomorowy (0,4%)
Infekcja: posocznica neutropeniczna (2,4%), posocznica (0,4%), odoskrzelowe zapalenie płuc (0,4%) Krew i
Limfatyczny: agranulocytoza (0,4%), zmniejszenie protrombiny (0,4%)
Naczyniowy: niedociśnienie (1,2%), żylne zapalenie żył i zakrzepowe zapalenie żył (0,4%), niedociśnienie ortostatyczne (0,8%)
Nerkowy: niewydolność nerek (0,4%)
Wątroby i dróg żółciowych: żółtaczka (0,4%), nieprawidłowe wyniki testów czynnościowych wątroby (0,4%), niewydolność wątroby (0,4%), śpiączka wątrobowa (0,4%), hepatotoksyczność (0,4%)
Układ odpornościowy: nadwrażliwość (1,2%)
Doświadczenie postmarketingowe
Następujące działania niepożądane obserwowano po wprowadzeniu produktu do obrotu: obrzęk naczynioruchowy, niewydolność wątroby, zwężenie przewodu łzowego, ostra niewydolność nerek wtórna do odwodnienia, w tym zgon [patrz OSTRZEŻENIA I ŚRODKI ], skórny toczeń rumieniowaty, choroby rogówki, w tym zapalenie rogówki, toksyczna leukoencefalopatia, ciężkie reakcje skórne, takie jak zespół Stevensa-Johnsona i toksyczna nekroliza naskórka (TEN) [patrz OSTRZEŻENIA I ŚRODKI ], uporczywy lub ciężki zespół dłoni i stóp może ostatecznie prowadzić do utraty odcisków palców [patrz OSTRZEŻENIA I ŚRODKI ]
W przypadku narażenia na rozkruszone tabletki XELODA 500 mg zgłaszano następujące działania niepożądane: podrażnienie i obrzęk oka, wysypka skórna, biegunka, parestezje, ból głowy, podrażnienie żołądka, wymioty i nudności.
INTERAKCJE Z LEKAMI
Interakcje między lekami
Antykoagulanty
U pacjentów przyjmujących XELODA 500 mg jednocześnie z lekami przeciwzakrzepowymi będącymi pochodnymi kumaryny, takimi jak warfaryna i fenprokumon [patrz PUDEŁKO OSTRZEŻENIE ]. Zdarzenia te wystąpiły w ciągu kilku dni i do kilku miesięcy po rozpoczęciu leczenia XELODA 500 mg, a w kilku przypadkach w ciągu 1 miesiąca po zaprzestaniu leczenia XELODA. Zdarzenia te wystąpiły u pacjentów z przerzutami do wątroby i bez przerzutów. W badaniu interakcji z pojedynczą dawką warfaryny stwierdzono znaczny wzrost średniej wartości AUC S-warfaryny [patrz FARMAKOLOGIA KLINICZNA ]. Maksymalna zaobserwowana wartość INR wzrosła o 91%. Ta interakcja jest prawdopodobnie spowodowana hamowaniem cytochromu P450 2C9 przez kapecytabinę i/lub jej metabolity.
Fenytoina
U pacjentów przyjmujących XELODA 500 mg należy dokładnie monitorować poziom fenytoiny i może być konieczne zmniejszenie dawki fenytoiny [patrz DAWKOWANIE I SPOSÓB PODAWANIA ]. Raporty po wprowadzeniu do obrotu wskazują, że niektórzy pacjenci otrzymujący XELODA 500 mg i fenytoinę mieli toksyczność związaną z podwyższonym poziomem fenytoiny. Nie przeprowadzono formalnych badań interakcji lekowych z fenytoiną, ale przypuszcza się, że mechanizm interakcji polega na hamowaniu izoenzymu CYP2C9 przez kapecytabinę i (lub) jej metabolity.
leukoworyna
Stężenie 5-fluorouracylu jest zwiększone, a jego toksyczność może być wzmocniona przez leukoworynę. U pacjentów w podeszłym wieku otrzymujących raz w tygodniu leukoworynę i fluorouracyl zgłaszano zgony z powodu ciężkiego zapalenia jelit, biegunki i odwodnienia.
Substraty CYP2C9
Poza warfaryną nie przeprowadzono formalnych badań dotyczących interakcji między produktem leczniczym XELODA a innymi substratami CYP2C9. Należy zachować ostrożność podczas jednoczesnego podawania produktu XELODA z substratami CYP2C9.
Allopurinol
Jednoczesne stosowanie z allopurynolem może zmniejszać stężenie aktywnych metabolitów kapecytabiny [patrz FARMAKOLOGIA KLINICZNA ], co może zmniejszać skuteczność leku XELODA. Należy unikać stosowania allopurynolu podczas leczenia lekiem XELODA.
Interakcja lek-pożywienie
Wykazano, że jedzenie zmniejsza zarówno szybkość, jak i stopień wchłaniania kapecytabiny [patrz FARMAKOLOGIA KLINICZNA ]. We wszystkich badaniach klinicznych pacjenci byli instruowani, aby podawać XELODA 500 mg w ciągu 30 minut po posiłku. Zaleca się podawanie produktu XELODA z pokarmem [patrz DAWKOWANIE I SPOSÓB PODAWANIA ].
OSTRZEŻENIA
Zawarte jako część "ŚRODKI OSTROŻNOŚCI" Sekcja
ŚRODKI OSTROŻNOŚCI
Koagulopatia
pacjentów otrzymujących jednocześnie kapecytabinę i doustne leki przeciwzakrzepowe z pochodnymi kumaryny należy ściśle monitorować odpowiedź przeciwzakrzepową (INR lub czas protrombinowy) z dużą częstotliwością, a dawkę leku przeciwzakrzepowego należy odpowiednio dostosować [patrz PUDEŁKO OSTRZEŻENIE oraz INTERAKCJE Z LEKAMI ].
Biegunka
XELODA 500mg może wywoływać biegunkę, czasem ciężką. Pacjenci z ciężką biegunką powinni być uważnie obserwowani i w przypadku odwodnienia powinni otrzymać uzupełnienie płynów i elektrolitów. U 875 pacjentów z rakiem piersi z przerzutami lub rakiem jelita grubego, którzy otrzymywali produkt XELODA w monoterapii, mediana czasu do pierwszego wystąpienia biegunki stopnia 2 do 4 wynosiła 34 dni (zakres od 1 do 369 dni). Mediana czasu trwania biegunki stopnia 3 do 4 wynosiła 5 dni. Biegunkę 2. stopnia w National Cancer Institute of Canada (NCIC) definiuje się jako wzrost od 4 do 6 stolców dziennie lub stolców nocnych, biegunkę 3. stopnia jako wzrost o 7 do 9 stolców na dobę lub nietrzymanie moczu i złe wchłanianie, a biegunkę 4. stopnia jako wzrost o ≥10 stolców/dobę lub rażąco krwawa biegunka lub potrzeba wsparcia pozajelitowego. W przypadku wystąpienia biegunki 2., 3. lub 4. stopnia, podawanie produktu XELODA należy natychmiast przerwać do czasu ustąpienia lub zmniejszenia nasilenia biegunki do stopnia 1. [patrz DAWKOWANIE I SPOSÓB PODAWANIA ]. Zaleca się standardowe leczenie przeciwbiegunkowe (np. loperamid).
Zgłaszano martwicze zapalenie jelit (zapalenie tyfusowe).
Kardiotoksyczność
Kardiotoksyczność obserwowana po zastosowaniu produktu XELODA 500 mg obejmuje zawał/niedokrwienie mięśnia sercowego, dusznicę bolesną, zaburzenia rytmu serca, zatrzymanie akcji serca, niewydolność serca, nagłą śmierć, zmiany elektrokardiograficzne i kardiomiopatię. Te działania niepożądane mogą występować częściej u pacjentów z chorobą wieńcową w wywiadzie.
Niedobór dehydrogenazy dihydropirymidynowej
Na podstawie doniesień postmarketingowych, pacjenci z pewnymi homozygotycznymi lub pewnymi złożonymi heterozygotycznymi mutacjami w genie DPD, które powodują całkowity lub prawie całkowity brak aktywności DPD, są narażeni na zwiększone ryzyko ostrej, wczesnej toksyczności i ciężkich, zagrażających życiu lub śmiertelnych działań niepożądanych. reakcje wywołane przez XELODA (np. zapalenie błon śluzowych, biegunka, neutropenia i neurotoksyczność). Pacjenci z częściową aktywnością DPD mogą również być narażeni na zwiększone ryzyko ciężkich, zagrażających życiu lub prowadzących do zgonu działań niepożądanych spowodowanych przez produkt XELODA.
Wstrzymać lub trwale odstawić XELODA 500 mg na podstawie oceny klinicznej początku, czasu trwania i nasilenia obserwowanej toksyczności u pacjentów z objawami ostrej, wczesnej lub niezwykle ciężkiej toksyczności, która może wskazywać na prawie całkowity lub całkowity brak aktywności DPD. Żadna dawka XELODA nie okazała się bezpieczna dla pacjentów z całkowitym brakiem aktywności DPD. Nie ma wystarczających danych, aby zalecić konkretną dawkę u pacjentów z częściową aktywnością DPD mierzoną dowolnym konkretnym testem.
Odwodnienie i niewydolność nerek
Obserwowano odwodnienie, które może powodować ostrą niewydolność nerek, która może być śmiertelna. Pacjenci z istniejącymi wcześniej zaburzeniami czynności nerek lub otrzymujący jednocześnie produkt XELODA ze znanymi środkami nefrotoksycznymi są narażeni na większe ryzyko. Pacjenci z anoreksją, osłabieniem, nudnościami, wymiotami lub biegunką mogą szybko się odwodnić. Monitoruj pacjentów podczas podawania produktu XELODA 500 mg, aby zapobiec odwodnieniu i skorygować je na początku. W przypadku wystąpienia odwodnienia stopnia 2. (lub wyższego) leczenie produktem XELODA należy natychmiast przerwać, a odwodnienie skorygować. Nie należy wznawiać leczenia, dopóki pacjent nie zostanie nawodniony, a wszelkie sprzyjające mu przyczyny nie zostaną usunięte lub opanowane. W razie potrzeby należy zmodyfikować dawkowanie w przypadku wystąpienia zdarzenia niepożądanego [patrz DAWKOWANIE I SPOSÓB PODAWANIA ].
Pacjenci z umiarkowanymi zaburzeniami czynności nerek na początku badania wymagają zmniejszenia dawki [patrz DAWKOWANIE I SPOSÓB PODAWANIA ]. Pacjenci z łagodnymi i umiarkowanymi zaburzeniami czynności nerek na początku leczenia powinni być uważnie obserwowani pod kątem wystąpienia działań niepożądanych. Jeśli u pacjenta wystąpi zdarzenie niepożądane stopnia 2 do 4, jak opisano w [patrz DAWKOWANIE I SPOSÓB PODAWANIA , Używaj w określonych populacjach , oraz FARMAKOLOGIA KLINICZNA ].
Toksyczność zarodkowo-płodowa
W oparciu o wyniki badań reprodukcji na zwierzętach i mechanizm działania, XELODA może powodować uszkodzenie płodu po podaniu kobiecie w ciąży [patrz FARMAKOLOGIA KLINICZNA ]. Ograniczone dostępne dane nie są wystarczające do poinformowania o stosowaniu leku XELODA 500 mg u kobiet w ciąży. W badaniach na zwierzętach dotyczących reprodukcji podawanie kapecytabiny ciężarnym zwierzętom w okresie organogenezy powodowało śmiertelność zarodków i działanie teratogenne u myszy oraz śmiertelność zarodków u małp przy odpowiednio 0,2 i 0,6-krotności ekspozycji (AUC) u pacjentów otrzymujących zalecaną dawkę [patrz Używaj w określonych populacjach ]. Poinformuj kobiety w ciąży o potencjalnym ryzyku dla płodu. Doradzić kobietom w wieku rozrodczym stosowanie skutecznej antykoncepcji podczas leczenia i przez 6 miesięcy po przyjęciu ostatniej dawki produktu XELODA [patrz Używaj w określonych populacjach ].
Toksyczność śluzówkowo-skórna i dermatologiczna
U pacjentów leczonych produktem XELODA mogą wystąpić ciężkie reakcje śluzówkowo-skórne, niektóre ze skutkiem śmiertelnym, takie jak zespół Stevensa-Johnsona i toksyczne martwicze oddzielanie się naskórka (TEN) [patrz DZIAŁANIA NIEPOŻĄDANE ]. Produkt XELODA 500 mg należy całkowicie odstawić u pacjentów, u których wystąpi ciężka reakcja śluzówkowo-skórna, prawdopodobnie związana z leczeniem produktem XELODA.
Zespół dłoniowo-podeszwowy (erytrodyzestezja dłoniowo-podeszwowa lub rumień akralny wywołany chemioterapią) jest toksycznym działaniem na skórę. Mediana czasu do wystąpienia wyniosła 79 dni (zakres od 11 do 360 dni) z zakresem nasilenia od 1 do 3 u pacjentów otrzymujących XELODA w monoterapii w dawce 500 mg w przypadku przerzutów. Stopień 1 charakteryzuje się którymkolwiek z następujących objawów: drętwienie, zaburzenia czucia/parestezje, mrowienie, bezbolesny obrzęk lub rumień rąk i (lub) stóp i (lub) dyskomfort niezakłócający normalnej aktywności. Zespół dłoniowo-podeszwowy stopnia 2. definiuje się jako bolesny rumień i obrzęk dłoni i (lub) stóp i (lub) dyskomfort zaburzający codzienną aktywność pacjenta. Zespół dłoniowo-stopowy stopnia 3. definiuje się jako wilgotne łuszczenie, owrzodzenie, powstawanie pęcherzy lub silny ból dłoni i (lub) stóp i (lub) silny dyskomfort, który uniemożliwia pacjentowi pracę lub wykonywanie codziennych czynności. Uporczywy lub ciężki zespół dłoni i stóp (stopień 2 i wyższy) może ostatecznie prowadzić do utraty odcisków palców, co może mieć wpływ na identyfikację pacjenta. W przypadku wystąpienia zespołu dłoniowo-podeszwowego stopnia 2. lub 3. podawanie produktu XELODA należy przerwać do czasu jego ustąpienia lub złagodzenia do stopnia 1. Po wystąpieniu zespołu dłoniowo-podeszwowego stopnia 3. kolejne dawki produktu XELODA należy zmniejszyć w dawce 500 mg [ Widzieć DAWKOWANIE I SPOSÓB PODAWANIA ].
Hiperbilirubinemia
875 pacjentów z rakiem piersi z przerzutami lub rakiem jelita grubego, którzy otrzymywali co najmniej jedną dawkę produktu XELODA 1250 mg/m2 dwa razy na dobę w monoterapii przez 2 tygodnie, po czym następował 1 tydzień przerwy, hiperbilirubinemia stopnia 3. (1,5-3 x GGN) wystąpiła u 875 pacjentów z rakiem piersi z przerzutami lub rakiem jelita grubego. 15,2% (n=133) pacjentów, a hiperbilirubinemia stopnia 4. (>3 x GGN) wystąpiła u 3,9% (n=34) pacjentów. Spośród 566 pacjentów z przerzutami do wątroby na początku badania i 309 pacjentów bez przerzutów do wątroby na początku badania hiperbilirubinemia stopnia 3 lub 4 wystąpiła odpowiednio u 22,8% i 12,3%. Spośród 167 pacjentów z hiperbilirubinemią stopnia 3 lub 4 u 18,6% (n=31) wystąpiło również podwyższenie aktywności fosfatazy zasadowej w okresie od 1 do 4 (stopnie od 1 do 4, bez podwyższenia na początku badania), a u 27,5% (n=46) podwyższenie aktywności aminotransferaz w dowolnym momencie (niekoniecznie jednocześnie). Większość z tych pacjentów, 64,5% (n=20) i 71,7% (n=33), miało przerzuty do wątroby na początku badania. Ponadto u 57,5% (n=96) i 35,3% (n=59) ze 167 pacjentów wystąpiło podwyższenie (stopnie od 1 do 4) zarówno przed rozpoczęciem, jak i po rozpoczęciu badania, odpowiednio fosfatazy alkalicznej lub aminotransferaz. Tylko 7,8% (n=13) i 3,0% (n=5) miało podwyższenie poziomu 3 lub 4 fosfatazy alkalicznej lub aminotransferaz.
596 pacjentów leczonych produktem XELODA 500 mg jako terapią pierwszego rzutu przerzutowego raka jelita grubego i odbytnicy częstość występowania hiperbilirubinemii stopnia 3 lub 4 była podobna do ogólnej bazy danych dotyczącej bezpieczeństwa stosowania produktu XELODA 500 mg w monoterapii w badaniu klinicznym. Mediana czasu do wystąpienia hiperbilirubinemii stopnia 3 lub 4 w populacji z rakiem jelita grubego wyniosła 64 dni, a mediana bilirubiny całkowitej wzrosła z 8 μm/l na początku do 13 μm/l podczas leczenia produktem XELODA. Spośród 136 pacjentów z rakiem jelita grubego z hiperbilirubinemią stopnia 3 lub 4, u 49 pacjentów wystąpiła hiperbilirubinemia stopnia 3 lub 4 jako ostatnia zmierzona wartość, z których 46 miało przerzuty do wątroby na początku badania.
U 251 pacjentek z rakiem piersi z przerzutami, które otrzymały skojarzenie produktu XELODA 500 mg i docetakselu, hiperbilirubinemia stopnia 3. (1,5 do 3 x GGN) wystąpiła u 7% (n=17), a hiperbilirubinemia stopnia 4. (>3 x GGN) wystąpiła u 2% (n=5).
przypadku wystąpienia związanego z lekiem zwiększenia stężenia bilirubiny stopnia 3 do 4, podawanie produktu XELODA 500 mg należy natychmiast przerwać do czasu, gdy hiperbilirubinemia zmniejszy się do ≤3,0 x GGN [patrz zalecane modyfikacje dawki w punkcie DAWKOWANIE I SPOSÓB PODAWANIA ].
Hematologiczny
U 875 pacjentów z rakiem piersi z przerzutami lub rakiem jelita grubego, którzy otrzymywali dawkę 1250 mg/m2 dwa razy na dobę w monoterapii przez 2 tygodnie, a następnie tygodniowy okres odpoczynku, 3,2%, 1,7% i 2,4% pacjentów miało stopień 3. lub 4 odpowiednio neutropenia, trombocytopenia lub zmniejszenie stężenia hemoglobiny. U 251 pacjentek z rakiem piersi z przerzutami, które otrzymały dawkę produktu XELODA w skojarzeniu z docetakselem, 68% miało neutropenię 3. lub 4. stopnia, 2,8% miało małopłytkowość 3. lub 4. stopnia, a 9,6% miało niedokrwistość 3. lub 4. stopnia.
Pacjenci z wyjściową liczbą neutrofili
Pacjenci geriatryczni
pacjentów w wieku ≥80 lat może wystąpić większa częstość występowania działań niepożądanych stopnia 3. lub 4. U 875 pacjentów z rakiem piersi z przerzutami lub rakiem jelita grubego, którzy otrzymywali XELODA 500 mg w monoterapii, u 62% z 21 pacjentów w wieku ≥80 lat leczonych produktem XELODA 500 mg wystąpiło związane z leczeniem zdarzenie niepożądane stopnia 3. lub 4.: biegunka u 6 osób (28,6%) , nudności u 3 (14,3%), zespół dłoniowo-stopowy u 3 (14,3%), a wymioty u 2 (9,5%) pacjentów. Wśród 10 pacjentów w wieku 70 lat i starszych (żaden pacjent nie był w wieku >80 lat) leczonych produktem XELODA 500 mg w skojarzeniu z docetakselem, 30% (3 z 10) pacjentów doświadczyło biegunki i zapalenia jamy ustnej stopnia 3. lub 4., a 40 % (4 z 10) doświadczyło zespołu dłoniowo-stopowego 3. stopnia.
Wśród 67 pacjentów w wieku ≥60 lat otrzymujących XELODA w skojarzeniu z docetakselem częstość występowania działań niepożądanych stopnia 3 lub 4 związanych z leczeniem, ciężkich działań niepożądanych związanych z leczeniem, wycofania z powodu działań niepożądanych, przerwania leczenia z powodu działań niepożądanych i leczenia przerwań w ciągu pierwszych dwóch cykli leczenia była większa niż w grupie pacjentów
995 pacjentów otrzymujących XELODA jako leczenie uzupełniające raka okrężnicy typu Dukes C po resekcji guza pierwotnego, u 41% z 398 pacjentów w wieku ≥65 lat leczonych produktem XELODA wystąpiło związane z leczeniem zdarzenie niepożądane stopnia 3. lub 4.: - zespół stopy u 75 (18,8%), biegunka u 52 (13,1%), zapalenie jamy ustnej u 12 (3,0%), neutropenia/granulocytopenia u 11 (2,8%), wymioty u 6 (1,5%) i nudności u 5 (1,3) %) pacjentów. U pacjentów w wieku ≥65 lat (cała randomizowana populacja; 188 pacjentów z kapecytabiną, 208 pacjentów z 5-FU/LV) leczonych z powodu raka okrężnicy typu Dukes C po resekcji guza pierwotnego, współczynniki ryzyka dla przeżycia wolnego od choroby i przeżycia całkowitego dla XELODA w porównaniu z 5-FU/LV wynosiły odpowiednio 1,01 (95% CI 0,80 – 1,27) i 1,04 (95% CI 0,79 – 1,37).
Niewydolność wątroby
Pacjenci z łagodnymi do umiarkowanych zaburzeniami czynności wątroby spowodowanymi przerzutami do wątroby powinni być uważnie obserwowani podczas podawania produktu XELODA. Wpływ ciężkich zaburzeń czynności wątroby na dyspozycję produktu XELODA nie jest znany [patrz Używaj w określonych populacjach oraz FARMAKOLOGIA KLINICZNA ].
Połączenie z innymi lekami
Stosowanie produktu XELODA w skojarzeniu z irynotekanem nie zostało odpowiednio zbadane.
Informacje dotyczące poradnictwa dla pacjentów
Poinformuj pacjenta, aby przeczytał zatwierdzone przez FDA oznakowanie pacjenta ( INFORMACJA O PACJENCIE ).
Biegunka
Należy poinformować pacjentów, u których występuje biegunka 2. stopnia (wzrost o 4 do 6 stolców na dobę lub stolce nocne) lub większa lub ciężka krwawa biegunka z silnym bólem brzucha i gorączką, aby przerwali stosowanie leku XELODA. Doradzić pacjentom stosowanie leków przeciwbiegunkowych (np. loperamidu) w celu opanowania biegunki [patrz OSTRZEŻENIA I ŚRODKI ].
Kardiotoksyczność
Poinformuj pacjentów o ryzyku kardiotoksyczności i natychmiast skontaktuj się z lekarzem lub udaj się na pogotowie ratunkowe w przypadku wystąpienia nowego bólu w klatce piersiowej, duszności, zawrotów głowy lub oszołomienia [patrz OSTRZEŻENIA I ŚRODKI ].
Niedobór dehydrogenazy dihydropirymidynowej
Doradź pacjentom, aby powiadomili swojego lekarza, jeśli mają znany niedobór DPD. Należy doradzić pacjentom, w przypadku całkowitego lub prawie całkowitego braku aktywności DPD, są oni narażeni na zwiększone ryzyko ostrej, wczesnej toksyczności i ciężkich, zagrażających życiu lub prowadzących do zgonu działań niepożądanych wywołanych przez XELODA (np. zapalenie błon śluzowych, biegunka, neutropenia i neurotoksyczność) [patrz OSTRZEŻENIA I ŚRODKI ].
Odwodnienie i niewydolność nerek
Należy poinstruować pacjentów, u których występuje odwodnienie stopnia 2. lub wyższego (wskazane płyny dożylne OSTRZEŻENIA I ŚRODKI ].
Ważne instrukcje administracyjne
Należy doradzić pacjentom połykanie tabletek XELODA 500 mg w całości, popijając wodą w ciągu 30 minut po posiłku. Należy doradzić pacjentom i opiekunom, aby nie kruszyli ani nie przecinali tabletek XELODA. Poradź pacjentom, jeśli nie mogą połykać tabletek XELODA w całości, aby poinformować swojego lekarza [patrz DAWKOWANIE I SPOSÓB PODAWANIA ].
Nadwrażliwość i obrzęk naczynioruchowy
Należy poinformować pacjentów, że XELODA może powodować ciężkie reakcje nadwrażliwości i obrzęk naczynioruchowy. Należy doradzić pacjentom, u których rozpoznano nadwrażliwość na kapecytabinę lub 5-fluorouracyl, poinformowanie ich lekarza [patrz PRZECIWWSKAZANIA ]. Należy poinstruować pacjentów, u których wystąpią reakcje nadwrażliwości lub objawy błon śluzowych (np. pokrzywka, wysypka, rumień, świąd lub obrzęk twarzy, warg, języka lub gardła, które utrudniają połykanie lub oddychanie), aby zaprzestali stosowania leku XELODA i natychmiast skontaktowali się z lekarzem. lub udać się na pogotowie. [Widzieć DZIAŁANIA NIEPOŻĄDANE ].
Mdłości
Należy poinstruować pacjentów, u których występują nudności stopnia 2. (znacznie zmniejszone spożycie pokarmu, ale mogące jeść z przerwami) lub większe, aby natychmiast przerwali przyjmowanie leku XELODA 500 mg i skontaktowali się z lekarzem w celu opanowania nudności [patrz DZIAŁANIA NIEPOŻĄDANE ].

Wymioty
Należy poinstruować pacjentów, u których wystąpią wymioty stopnia 2. (2 do 5 epizodów w ciągu 24 godzin) lub wyższego, aby natychmiast przerwali przyjmowanie leku XELODA i skontaktowali się z lekarzem w celu leczenia wymiotów [patrz DZIAŁANIA NIEPOŻĄDANE ].
Zespół dłoni i stóp
Należy poinstruować pacjentów, u których występuje zespół dłoniowo-podeszwowy stopnia 2. (bolesny rumień i obrzęk rąk i (lub) stóp i (lub) dyskomfort wpływające na codzienne czynności pacjenta) lub wyższego, aby natychmiast przerwali przyjmowanie leku XELODA 500 mg i skontaktowali się z lekarzem . Poinformuj pacjentów, że zalecane jest rozpoczęcie leczenia objawowego, a zespół dłoni i stóp może prowadzić do utraty odcisków palców, co może mieć wpływ na osobistą identyfikację [patrz DZIAŁANIA NIEPOŻĄDANE ].
Zapalenie jamy ustnej
Należy poinformować pacjentów, u których występuje zapalenie jamy ustnej stopnia 2. (bolesny rumień, obrzęk lub owrzodzenie jamy ustnej lub języka, ale z możliwością jedzenia) lub wyższego, aby natychmiast przerwali przyjmowanie leku XELODA i skontaktowali się z lekarzem [patrz DZIAŁANIA NIEPOŻĄDANE ].
Gorączka i neutropenia
Poinformuj pacjentów, u których wystąpi gorączka 100,5°F lub wyższa lub inne oznaki potencjalnej infekcji, aby skontaktowali się z lekarzem [patrz DZIAŁANIA NIEPOŻĄDANE ].
Toksyczność zarodkowo-płodowa
Poinformuj kobiety o potencjale rozrodczym, potencjalnym ryzyku dla płodu oraz o stosowaniu skutecznej antykoncepcji podczas leczenia produktem XELODA 500 mg i przez 6 miesięcy po podaniu ostatniej dawki. Doradź kobietom, aby poinformowały swojego lekarza o stwierdzonej lub podejrzewanej ciąży [patrz OSTRZEŻENIA I ŚRODKI , Używaj w określonych populacjach ].
Należy doradzić pacjentom płci męskiej mającym partnerki w wieku rozrodczym stosowanie skutecznej antykoncepcji podczas leczenia preparatem XELODA 500 mg i przez 3 miesiące po przyjęciu ostatniej dawki [patrz Używaj w określonych populacjach ].
Laktacja
Doradzać kobietom, aby nie karmiły piersią podczas leczenia lekiem XELODA 500 mg i przez 2 tygodnie po przyjęciu ostatniej dawki [patrz Używaj w określonych populacjach ].
Toksykologia niekliniczna
rakotwórczość, mutageneza, upośledzenie płodności
Nie przeprowadzono odpowiednich badań oceniających rakotwórczy potencjał kapecytabiny. Kapecytabina nie wykazywała działania mutagennego in vitro na bakterie (test Amesa) lub komórki ssaków (test mutacji genu V79/HPRT chomika chińskiego). Kapecytabina działała klastogennie in vitro na ludzkie limfocyty krwi obwodowej, ale nie działała klastogennie in vivo na szpik kostny myszy (test mikrojądrowy). Fluorouracyl powoduje mutacje w bakteriach i drożdżach. Fluorouracyl powoduje również nieprawidłowości chromosomalne w mysim teście mikrojądrowym in vivo.
badaniach płodności i ogólnej sprawności rozrodczej samic myszy doustna dawka kapecytabiny wynosząca 760 mg/kg/dzień (około 2300 mg/m2/dzień) zaburzała ruję i w konsekwencji powodowała spadek płodności. U myszy, które zaszły w ciążę, żaden płód nie przeżył tej dawki. Zaburzenie rui było odwracalne. U mężczyzn dawka ta powodowała zmiany zwyrodnieniowe w jądrach, w tym zmniejszenie liczby spermatocytów i spermatyd. W oddzielnych badaniach farmakokinetycznych ta dawka u myszy dawała wartości AUC 5'-DFUR około 0,7 razy większe od odpowiednich wartości u pacjentów otrzymujących zalecaną dawkę dobową.
Używaj w określonych populacjach
Ciąża
Podsumowanie ryzyka
W oparciu o wyniki badań rozrodu zwierząt i mechanizm działania, XELODA 500 mg może powodować uszkodzenie płodu po podaniu kobiecie w ciąży [patrz FARMAKOLOGIA KLINICZNA ]. Ograniczone dostępne dane dotyczące ludzi nie są wystarczające do poinformowania o ryzyku związanym z lekiem w czasie ciąży. W badaniach na zwierzętach dotyczących reprodukcji podawanie kapecytabiny ciężarnym zwierzętom w okresie organogenezy powodowało śmiertelność zarodków i działanie teratogenne u myszy oraz śmiertelność zarodków u małp przy odpowiednio 0,2 i 0,6-krotności ekspozycji (AUC) u pacjentów otrzymujących zalecaną dawkę [patrz Dane ]. Poinformuj kobiety w ciąży o potencjalnym ryzyku dla płodu.
Szacowane ryzyko tła poważnych wad wrodzonych i poronienia dla wskazanej populacji jest nieznane. W ogólnej populacji USA szacowane podstawowe ryzyko poważnych wad wrodzonych i poronienia w klinicznie rozpoznanych ciążach wynosi odpowiednio 2-4% i 15-20%.
Dane
Dane zwierząt
Doustne podawanie kapecytabiny ciężarnym myszom w okresie organogenezy w dawce 198 mg/kg/dobę powodowało wady rozwojowe i śmiertelność zarodków. W oddzielnych badaniach farmakokinetycznych ta dawka u myszy dawała wartości AUC 5'-DFUR, które były około 0,2 razy większe od wartości AUC u pacjentów, którym podawano zalecaną dawkę dobową. Wady rozwojowe u myszy obejmowały rozszczep podniebienia, brak oka, mikroftalmię, oligodaktylię, polidaktylię, syndaktylię, skręcony ogon i rozszerzenie komór mózgowych. Doustne podawanie kapecytabiny ciężarnym małpom w okresie organogenezy w dawce 90 mg/kg/dobę powodowało śmiertelność płodów. Ta dawka dawała wartości AUC 5'-DFUR, które były około 0,6 razy większe od wartości AUC u pacjentów otrzymujących zalecaną dawkę dobową.
Laktacja
Podsumowanie ryzyka
Nie ma informacji dotyczących obecności kapecytabiny w mleku kobiecym ani jej wpływu na produkcję mleka u niemowląt karmionych piersią. Metabolity kapecytabiny były obecne w mleku karmiących myszy [patrz Dane ]. Ze względu na możliwość wystąpienia ciężkich działań niepożądanych związanych z narażeniem na kapecytabinę u niemowląt karmionych piersią należy odradzać karmienie piersią podczas leczenia produktem XELODA 500 mg i przez 2 tygodnie po przyjęciu ostatniej dawki.
Dane
Myszy karmiące, którym podano pojedynczą doustną dawkę kapecytabiny, wydalały do mleka znaczne ilości metabolitów kapecytabiny.
Kobiety i mężczyźni o potencjale rozrodczym
Testy ciążowe
Testy ciążowe są zalecane u samic w wieku rozrodczym przed rozpoczęciem stosowania produktu XELODA.
Zapobieganie ciąży
Kobiety
XELODA 500 mg może powodować uszkodzenie płodu po podaniu kobiecie w ciąży [patrz Ciąża ]. Doradzić kobietom w wieku rozrodczym stosowanie skutecznej antykoncepcji podczas leczenia i przez 6 miesięcy po przyjęciu ostatniej dawki produktu XELODA.
Mężczyźni
W oparciu o wyniki badań toksyczności genetycznej należy doradzić pacjentom płci męskiej mającym partnerki w wieku rozrodczym stosowanie skutecznej antykoncepcji podczas leczenia i przez 3 miesiące po przyjęciu ostatniej dawki produktu XELODA [patrz Toksykologia niekliniczna ].
Bezpłodność
Na podstawie badań na zwierzętach XELODA może zaburzać płodność u samic i samców w wieku rozrodczym [patrz Toksykologia niekliniczna ].
Zastosowanie pediatryczne
Nie ustalono bezpieczeństwa i skuteczności produktu XELODA u dzieci. Nie wykazano korzyści klinicznych w dwóch badaniach jednoramiennych u dzieci z nowo rozpoznanymi glejakami pnia mózgu i glejakami wysokiego stopnia złośliwości. W obu badaniach pacjenci pediatryczni otrzymywali eksperymentalną postać pediatryczną kapecytabiny jednocześnie z i po zakończeniu radioterapii (całkowita dawka 5580 cGy we frakcjach 180 cGy). Względna biodostępność preparatu badanego do preparatu XELODA 500 mg była podobna.
Pierwsze badanie przeprowadzono u 22 pacjentów pediatrycznych (mediana wieku 8 lat, zakres 5-17 lat) z nowo zdiagnozowanymi nierozsianymi glejakami wewnątrzpochodnymi pnia mózgu i glejakami wysokiego stopnia zaawansowania. W części badania dotyczącej ustalania dawki pacjenci otrzymywali kapecytabinę z towarzyszącą radioterapią w dawkach od 500 mg/m2 do 850 mg/m2 co 12 godzin przez okres do 9 tygodni. Po dwutygodniowej przerwie pacjenci otrzymywali 1250 mg/m2 kapecytabiny co 12 godzin w dniach 1-14 21-dniowego cyklu przez maksymalnie 3 cykle. Maksymalna tolerowana dawka (MTD) kapecytabiny podawana jednocześnie z radioterapią wynosiła 650 mg/m2 co 12 godzin. Głównymi działaniami toksycznymi ograniczającymi dawkę były erytrodyzestezja dłoniowo-podeszwowa i zwiększenie aktywności aminotransferazy alaninowej (ALT).
Drugie badanie przeprowadzono u 34 dodatkowych pacjentów pediatrycznych z nowo zdiagnozowanymi nierozsianymi glejakami wewnątrzpochodnymi pnia mózgu (mediana wieku 7 lat, zakres 3-16 lat) i 10 pacjentów pediatrycznych, którzy otrzymywali MTD kapecytabiny w kryteria kwalifikowalności do tego badania. Wszyscy pacjenci otrzymywali 650 mg/m2 kapecytabiny co 12 godzin z jednoczesną radioterapią przez okres do 9 tygodni. Po dwutygodniowej przerwie pacjenci otrzymywali 1250 mg/m2 kapecytabiny co 12 godzin w dniach 1-14 21-dniowego cyklu przez maksymalnie 3 cykle.
Nie stwierdzono poprawy rocznego wskaźnika przeżycia bez progresji i rocznego wskaźnika przeżycia całkowitego u dzieci z nowo rozpoznanymi glejakami pnia mózgu, którzy otrzymywali kapecytabinę, w porównaniu z podobną populacją pacjentów pediatrycznych, którzy uczestniczyli w innych badaniach klinicznych.
Profil działań niepożądanych kapecytabiny był zgodny ze znanym profilem działań niepożądanych u dorosłych, z wyjątkiem nieprawidłowości laboratoryjnych, które występowały częściej u dzieci. Najczęściej zgłaszanymi nieprawidłowościami laboratoryjnymi (częstość występowania ≥40%) była zwiększona aktywność AlAT (75%), limfocytopenia (73%), leukopenia (73%), hipokaliemia (68%), małopłytkowość (57%), hipoalbuminemia (55). %), neutropenia (50%), niski hematokryt (50%), hipokalcemia (48%), hipofosfatemia (45%) i hiponatremia (45%).
Zastosowanie geriatryczne
Lekarze powinni zwracać szczególną uwagę na monitorowanie działań niepożądanych XELODA 500mg u osób starszych [patrz OSTRZEŻENIA I ŚRODKI ].
Niewydolność wątroby
Należy zachować ostrożność podczas leczenia produktem XELODA pacjentów z łagodnymi do umiarkowanych zaburzeniami czynności wątroby spowodowanymi przerzutami do wątroby. Wpływ ciężkich zaburzeń czynności wątroby na produkt XELODA nie jest znany [patrz OSTRZEŻENIA I ŚRODKI oraz FARMAKOLOGIA KLINICZNA ].
Niewydolność nerek
Pacjenci z umiarkowanymi (klirens kreatyniny = 30 do 50 ml/min) i ciężkimi (klirens kreatyniny PRZECIWWSKAZANIA , OSTRZEŻENIA I ŚRODKI , DAWKOWANIE I SPOSÓB PODAWANIA , oraz FARMAKOLOGIA KLINICZNA ].
PRZEDAWKOWAĆ
Do objawów ostrego przedawkowania należą nudności, wymioty, biegunka, podrażnienie i krwawienie z przewodu pokarmowego oraz zahamowanie czynności szpiku kostnego. Postępowanie medyczne w przypadku przedawkowania powinno obejmować zwyczajowe wspomagające interwencje medyczne mające na celu skorygowanie występujących objawów klinicznych. Chociaż nie zgłoszono żadnego doświadczenia klinicznego z zastosowaniem dializy w leczeniu przedawkowania produktu XELODA 500 mg, dializa może przynieść korzyści w zakresie zmniejszenia stężenia 5'-DFUR, metabolitu związku macierzystego o małej masie cząsteczkowej.
Pojedyncze dawki produktu XELODA 500 mg nie były śmiertelne dla myszy, szczurów i małp w dawkach do 2000 mg/kg (2,4, 4,8 i 9,6-krotność zalecanej dawki dobowej dla ludzi w przeliczeniu na mg/m2).
PRZECIWWSKAZANIA
Ciężka niewydolność nerek
XELODA jest przeciwwskazany u pacjentów z ciężkimi zaburzeniami czynności nerek (klirens kreatyniny poniżej 30 ml/min [Cockroft i Gault]) [patrz Używaj w określonych populacjach oraz FARMAKOLOGIA KLINICZNA ].
Nadwrażliwość
XELODA 500mg jest przeciwwskazany u pacjentów ze stwierdzoną nadwrażliwością na kapecytabinę lub którykolwiek z jej składników. XELODA jest przeciwwskazany u pacjentów ze znaną nadwrażliwością na 5-fluorouracyl.
FARMAKOLOGIA KLINICZNA
Mechanizm akcji
Enzymy przekształcają kapecytabinę w 5-fluorouracyl (5-FU) in vivo. Zarówno komórki zdrowe, jak i nowotworowe metabolizują 5-FU do 5-fluoro-2'-deoksyurydyno-monofosforanu (FdUMP) i 5-fluorourydynotrifosforanu (FUTP). Te metabolity powodują uszkodzenie komórek przez dwa różne mechanizmy. Po pierwsze, FdUMP i kofaktor kwasu foliowego, N5-10-metylenotetrahydrofolian, wiążą się z syntazą tymidylanową (TS), tworząc kowalencyjnie związany kompleks trójskładnikowy. Wiązanie to hamuje tworzenie tymidylanu z 2'-dezoksyurydylanu. Tymidylan jest niezbędnym prekursorem trifosforanu tymidyny, który jest niezbędny do syntezy DNA, dzięki czemu niedobór tego związku może hamować podział komórek. Po drugie, jądrowe enzymy transkrypcyjne mogą błędnie włączać FUTP w miejsce trifosforanu urydyny (UTP) podczas syntezy RNA. Ten błąd metaboliczny może zakłócać przetwarzanie RNA i syntezę białek.
Farmakokinetyka
Wchłanianie
Po podaniu doustnym 1255 mg/m2 dwa razy na dobę pacjentom z nowotworami, kapecytabina osiągnęła maksymalne stężenie we krwi po około 1,5 godziny (Tmax), przy czym maksymalne stężenie 5-FU wystąpiło nieco później, po 2 godzinach. Pokarm zmniejszał zarówno szybkość, jak i stopień wchłaniania kapecytabiny, przy czym średnie Cmax i AUC0-∞ zmniejszyły się odpowiednio o 60% i 35%. Cmax i AUC0-∞ 5-FU były również zmniejszone przez jedzenie odpowiednio o 43% i 21%. Tmax opóźniony przez pokarm dla obu rodziców i 5-FU o 1,5 godziny [patrz OSTRZEŻENIA I ŚRODKI , DAWKOWANIE I SPOSÓB PODAWANIA , oraz Interakcja lek-pożywienie ].
Farmakokinetykę XELODA 500 mg i jej metabolitów oceniano u około 200 pacjentów z rakiem w zakresie dawek od 500 do 3500 mg/m2/dobę. W tym zakresie farmakokinetyka produktu XELODA i jego metabolitu 5'-DFCR była proporcjonalna do dawki i nie zmieniała się w czasie. Jednak wzrosty AUC 5'-DFUR i 5-FU były większe niż proporcjonalne do zwiększenia dawki, a AUC 5-FU było o 34% większe w 14. dniu niż w 1. dniu. Cmax i AUC 5-FU były większe niż 85%.
Dystrybucja
Wiązanie kapecytabiny i jej metabolitów z białkami osocza wynosi mniej niż 60% i nie jest zależne od stężenia. Kapecytabina wiązała się głównie z ludzką albuminą (około 35%). XELODA 500 mg ma niski potencjał interakcji farmakokinetycznych związanych z wiązaniem z białkami osocza.
Bioaktywacja i metabolizm
Kapecytabina jest intensywnie metabolizowana enzymatycznie do 5-FU. W wątrobie karboksyloesteraza o masie 60 kDa hydrolizuje większość związku do 5'-deoksy-5-fluorocytydyny (5'-DFCR). Deaminaza cytydynowa, enzym występujący w większości tkanek, w tym w nowotworach, następnie przekształca 5'-DFCR w 5'-DFUR. Enzym, fosforylaza tymidynowa (dThdPaza), następnie hydrolizuje 5'DFUR do aktywnego leku 5-FU. Wiele tkanek w całym ciele wyraża fosforylazę tymidynową. Niektóre ludzkie nowotwory wykazują ekspresję tego enzymu w wyższych stężeniach niż otaczające zdrowe tkanki. Po doustnym podaniu produktu XELODA na 7 dni przed zabiegiem chirurgicznym pacjentom z rakiem jelita grubego średni stosunek stężenia 5-FU w guzach jelita grubego do przylegających tkanek wynosił 2,9 (zakres od 0,9 do 8,0). Te proporcje nie były oceniane u pacjentów z rakiem piersi ani w porównaniu z infuzją 5-FU.
Szlak metaboliczny kapecytabiny do 5-FU
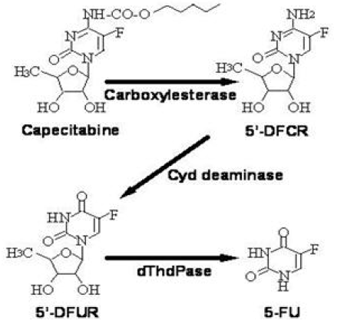
Enzym dehydrogenaza dihydropirymidynowa uwodornia 5-FU, produkt metabolizmu kapecytabiny, do znacznie mniej toksycznego 5-fluoro-5,6-dihydro-fluorouracylu (FUH2). Dihydropirymidynaza rozszczepia pierścień pirymidynowy z wytworzeniem kwasu 5-fluoroureidopropionowego (FUPA). Wreszcie β-ureido-propionaza rozszczepia FUPA do α-fluoro-β-alaniny (FBAL), która jest usuwana z moczem.
Badania enzymatyczne in vitro na mikrosomach wątroby ludzkiej wykazały, że kapecytabina i jej metabolity (5'-DFUR, 5'-DFCR, 5-FU i FBAL) nie hamują metabolizmu badanych substratów przez izoenzymy 1A2, 2A6, 3A4 cytochromu P450. 2C19, 2D6 i 2E1.
Wydalanie
Kapecytabina i jej metabolity są wydalane głównie z moczem; 95,5% podanej dawki kapecytabiny jest wydalane z moczem. Wydalanie z kałem jest minimalne (2,6%). Głównym metabolitem wydalanym z moczem jest FBAL, który stanowi 57% podanej dawki. Około 3% podanej dawki jest wydalane z moczem w postaci niezmienionej. Okres półtrwania w fazie eliminacji zarówno macierzystej kapecytabiny, jak i 5-FU wynosił około 0,75 godziny.
Wpływ wieku, płci i rasy na farmakokinetykę kapecytabiny
Analiza populacji zbiorczych danych z dwóch dużych kontrolowanych badań u pacjentów z przerzutowym rakiem jelita grubego (n=505), którym podawano XELODA 500 mg w dawce 1250 mg/m2 dwa razy na dobę, wykazała, że płeć (202 kobiety i 303 mężczyźni) oraz rasa (455) pacjentów rasy białej/kaukaskiej, 22 pacjentów rasy czarnej i 28 pacjentów innej rasy) nie mają wpływu na farmakokinetykę 5'DFUR, 5-FU i FBAL. Wiek nie ma znaczącego wpływu na farmakokinetykę 5'-DFUR i 5-FU w okresie od 27 do 86 lat. 20% wzrost wieku powoduje 15% wzrost AUC FBAL [patrz OSTRZEŻENIA I ŚRODKI oraz DAWKOWANIE I SPOSÓB PODAWANIA ].
Po podaniu doustnym 825 mg/m2 kapecytabiny dwa razy na dobę przez 14 dni pacjenci z Japonii (n=18) mieli około 36% niższe Cmax i 24% niższe AUC dla kapecytabiny niż pacjenci rasy kaukaskiej (n=22). Pacjenci japońscy mieli również około 25% niższe Cmax i 34% niższe AUC dla FBAL niż pacjenci rasy kaukaskiej. Kliniczne znaczenie tych różnic nie jest znane. Nie wystąpiły istotne różnice w ekspozycji na inne metabolity (5'-DFCR, 5'-DFUR i 5FU).
Efekt niewydolności wątroby
XELODA oceniano u 13 pacjentów z łagodnymi do umiarkowanych zaburzeniami czynności wątroby spowodowanymi przerzutami do wątroby zdefiniowanymi na podstawie łącznej punktacji obejmującej bilirubinę, AspAT/AlAT i fosfatazę alkaliczną po podaniu pojedynczej dawki 1255 mg/m2 produktu XELODA. Zarówno AUC0-∞, jak i Cmax kapecytabiny wzrosły o 60% u pacjentów z zaburzeniami czynności wątroby w porównaniu z pacjentami z prawidłową czynnością wątroby (n=14). AUC0-∞ i Cmax 5-FU nie uległy zmianie. U pacjentów z łagodnymi do umiarkowanych zaburzeniami czynności wątroby spowodowanymi przerzutami do wątroby należy zachować ostrożność podczas podawania produktu XELODA 500 mg. Wpływ ciężkich zaburzeń czynności wątroby na XELODA 500 mg nie jest znany [patrz OSTRZEŻENIA I ŚRODKI oraz Używaj w specjalnych populacjach ].
Skutek niewydolności nerek
Po doustnym podaniu kapecytabiny w dawce 1250 mg/m2 dwa razy na dobę pacjentom z nowotworami i różnym stopniem niewydolności nerek, u pacjentów z umiarkowaną (klirens kreatyniny = 30 do 50 ml/min) i ciężką (klirens kreatyniny 80 ml/min). Ekspozycja ogólnoustrojowa na 5'-DFUR była odpowiednio o 42% i 71% większa u pacjentów z umiarkowaną i ciężką niewydolnością nerek niż u zdrowych pacjentów. Ekspozycja ogólnoustrojowa na kapecytabinę była o około 25% większa zarówno u pacjentów z umiarkowaną, jak i ciężką niewydolnością nerek [patrz DAWKOWANIE I SPOSÓB PODAWANIA , PRZECIWWSKAZANIA , OSTRZEŻENIA I ŚRODKI , oraz Używaj w specjalnych populacjach ].
Wpływ kapecytabiny na farmakokinetykę warfaryny
czterech pacjentów z nowotworem przewlekłe podawanie kapecytabiny (1250 mg/m2 dwa razy na dobę) z pojedynczą dawką 20 mg warfaryny zwiększyło średnią wartość AUC S-warfaryny o 57% i zmniejszyło jej klirens o 37%. Skorygowana początkowa wartość AUC INR u tych 4 pacjentów wzrosła 2,8-krotnie, a maksymalna obserwowana średnia wartość INR wzrosła o 91% [patrz PUDEŁKO OSTRZEŻENIE oraz INTERAKCJE Z LEKAMI ].
Wpływ leków zobojętniających kwas na farmakokinetykę kapecytabiny
Gdy Maalox® (20 ml), środek zobojętniający kwas zawierający wodorotlenek glinu i wodorotlenek magnezu, podano bezpośrednio po podaniu produktu XELODA (1250 mg/m2, n=12 pacjentów z rakiem), AUC i Cmax wzrosły odpowiednio o 16% i 35%, dla kapecytabiny i odpowiednio o 18% i 22% dla 5'-DFCR. Nie zaobserwowano wpływu na pozostałe trzy główne metabolity (5'-DFUR, 5-FU, FBAL) preparatu XELODA.
Wpływ allopurynolu na kapecytabinę
opublikowanym piśmiennictwie doniesiono, że jednoczesne stosowanie z allopurynolem może zmniejszać konwersję kapecytabiny do aktywnych metabolitów, FdUMP i FUTP; jednakże znaczenie kliniczne nie zostało w pełni scharakteryzowane.
Wpływ kapecytabiny na farmakokinetykę docetakselu i odwrotnie
Badanie fazy 1 oceniało wpływ preparatu XELODA na farmakokinetykę docetakselu (Taxotere®) oraz wpływ docetakselu na farmakokinetykę XELODA u 26 pacjentów z guzami litymi. Stwierdzono, że XELODA nie ma wpływu na farmakokinetykę docetakselu (Cmax i AUC), a docetaksel nie ma wpływu na farmakokinetykę kapecytabiny i prekursora 5-FU 5'-DFUR.
Studia kliniczne
Adiuwantowy rak okrężnicy
Wieloośrodkowe, randomizowane, kontrolowane badanie kliniczne III fazy z udziałem pacjentów z rakiem okrężnicy typu Dukes C (X-ACT) dostarczyło danych dotyczących stosowania produktu XELODA w leczeniu uzupełniającym pacjentów z rakiem okrężnicy. Podstawowym celem badania było porównanie przeżycia wolnego od choroby (DFS) u pacjentów otrzymujących XELODA z pacjentami otrzymującymi wyłącznie dożylnie 5-FU/LV. W tym badaniu 1987 pacjentów zostało losowo przydzielonych do leczenia produktem XELODA 1250 mg/m2 doustnie dwa razy na dobę przez 2 tygodnie, a następnie do 1-tygodniowego okresu odpoczynku, podawanego jako 3-tygodniowe cykle łącznie 8 cykli (24 tygodnie) lub i.v. bolus 5-FU 425 mg/m2 i 20 mg/m2 dożylnie leukoworyny w dniach od 1 do 5, podawane jako 4-tygodniowe cykle, łącznie 6 cykli (24 tygodnie). Pacjenci biorący udział w badaniu musieli być w wieku od 18 do 75 lat z histologicznie potwierdzonym rakiem okrężnicy w stopniu zaawansowania C według skali Dukesa z co najmniej jednym dodatnim węzłem chłonnym i przejść (w ciągu 8 tygodni przed randomizacją) całkowitą resekcję guza pierwotnego bez makroskopowych lub mikroskopowych dowodów pozostałego guza. Pacjenci musieli również nie mieć wcześniejszej chemioterapii lub immunoterapii cytotoksycznej (z wyjątkiem sterydów) i mieć stan wydolności ECOG 0 lub 1 (KPS ≥ 70%), ANC ≥ 1,5x109/l, płytki krwi ≥ 100x109/l, stężenie kreatyniny w surowicy ≤ 1,5 GGN, bilirubina całkowita ≤ 1,5 GGN, AspAT/AlAT ≤ 2,5 GGN i CEA w granicach normy w momencie randomizacji.
Wyjściowe dane demograficzne pacjentów z grupy XELODA i 5-FU/LV przedstawiono w Tabeli 10. Wyjściowa charakterystyka była dobrze zrównoważona między ramionami.
Wszyscy pacjenci z prawidłową czynnością nerek lub łagodnymi zaburzeniami czynności nerek rozpoczęli leczenie pełną dawką początkową 1250 mg/m2 pc. doustnie dwa razy na dobę. Dawka początkowa została zmniejszona u pacjentów z umiarkowanymi zaburzeniami czynności nerek (obliczony klirens kreatyniny od 30 do 50 ml/min) na początku badania [patrz DAWKOWANIE I SPOSÓB PODAWANIA ]. Następnie, dla wszystkich pacjentów, dawki były dostosowywane w razie potrzeby w zależności od toksyczności. Zarządzanie dawką produktu XELODA 500 mg obejmowało zmniejszenie dawki, opóźnienie cyklu i przerwanie leczenia (patrz Tabela 11).
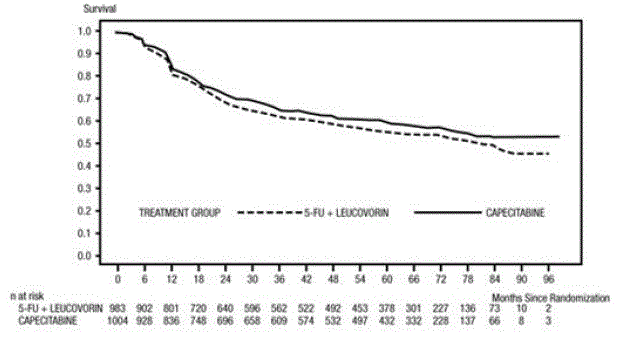
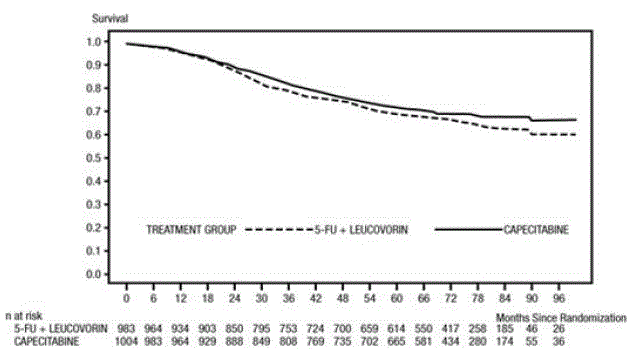
Mediana czasu obserwacji w momencie analizy wyniosła 83 miesiące (6,9 roku). Współczynnik ryzyka dla DFS dla produktu XELODA 500 mg w porównaniu z 5-FU/LV wyniósł 0,88 (95% CI 0,77 – 1,01) (patrz i Rysunek 1 ). Ponieważ górna dwustronna granica ufności 95% współczynnika ryzyka była mniejsza niż 1,20, XELODA nie była gorsza niż 5-FU/LV. Wybór marginesu non-inferiority wynoszący 1,20 odpowiada zachowaniu około 75% wpływu 5-FU/LV na DFS. Współczynnik ryzyka dla XELODA w porównaniu z 5-FU/LV w odniesieniu do całkowitego przeżycia wyniósł 0,86 (95% CI 0,74 – 1,01). Wskaźnik 5-letniego przeżycia całkowitego wyniósł 71,4% dla XELODA i 68,4% dla 5-FU/LV (patrz Rycina 2).
Rycina 1 Szacunki Kaplana-Meiera dotyczące przeżycia wolnego od choroby (cała populacja randomizowana)a
Rycina 2 Szacunki Kaplana-Meiera dotyczące całkowitego przeżycia (cała populacja zrandomizowana)
Przerzutowy rak jelita grubego
Ogólny
Zalecana dawka XELODA 500mg została określona w otwartym, randomizowanym badaniu klinicznym, badającym skuteczność i bezpieczeństwo ciągłej terapii kapecytabiną (1331 mg/m2/dobę w dwóch dawkach podzielonych, n=39), przerywanej terapii kapecytabiną ( 2510 mg/m2/dobę w dwóch dawkach podzielonych, n=34) oraz leczenie przerywane kapecytabiną w skojarzeniu z leukoworyną doustną (LV) (kapecytabina 1657 mg/m2/dobę w dwóch dawkach podzielonych, n=35; leukoworyna 60 mg/ dobowej) u pacjentów z zaawansowanym i/lub przerzutowym rakiem jelita grubego w pierwszej linii z przerzutami. Nie było widocznej przewagi w odsetku odpowiedzi po dodaniu leukoworyny do produktu XELODA; jednak toksyczność wzrosła. XELODA w dawce 1250 mg/m2 dwa razy na dobę przez 14 dni, po której następował 1 tydzień przerwy, została wybrana do dalszego rozwoju klinicznego na podstawie ogólnego profilu bezpieczeństwa i skuteczności trzech badanych schematów.
Monoterapia
Dane z dwóch otwartych, wieloośrodkowych, randomizowanych, kontrolowanych badań klinicznych z udziałem 1207 pacjentów potwierdzają zastosowanie produktu XELODA w leczeniu pierwszego rzutu pacjentów z przerzutowym rakiem jelita grubego. Oba badania kliniczne miały identyczny projekt i zostały przeprowadzone w 120 ośrodkach w różnych krajach. Badanie 1 przeprowadzono w USA, Kanadzie, Meksyku i Brazylii; Badanie 2 przeprowadzono w Europie, Izraelu, Australii, Nowej Zelandii i na Tajwanie. Łącznie w obu badaniach 603 pacjentów zostało losowo przydzielonych do leczenia produktem XELODA 500 mg w dawce 1250 mg/m2 dwa razy na dobę przez 2 tygodnie, po czym następował 1-tygodniowy okres przerwy i podawany był w cyklach 3-tygodniowych; 604 pacjentów przydzielono losowo do leczenia 5-FU i leukoworyną (20 mg/m2 leukoworyny dożylnie, a następnie 425 mg/m2 dożylnie bolus 5-FU, w dniach od 1 do 5, co 28 dni).
obu badaniach oceniano przeżycie całkowite, czas do progresji i odsetek odpowiedzi (odpowiedzi pełne i częściowe). Odpowiedzi zostały określone przez kryteria Światowej Organizacji Zdrowia i przesłane do zaślepionej niezależnej komisji oceniającej (IRC). Różnice w ocenach między badaczem a IRC zostały uzgodnione przez sponsora, który nie znał ramienia leczenia, zgodnie z określonym algorytmem. Przeżycie oceniano na podstawie analizy non-inferiority.
Wyjściowe dane demograficzne pacjentów z grupy XELODA i 5-FU/LV przedstawiono w Tabeli 13.
Punkty końcowe skuteczności dla dwóch badań fazy 3 przedstawiono w i
Rycina 3 Krzywa Kaplana-Meiera dla całkowitego przeżycia połączonych danych (Badania 1 i 2)
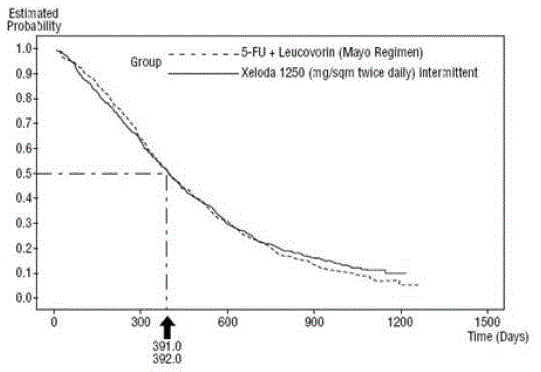
XELODA był lepszy od 5-FU/LV pod względem odsetka obiektywnych odpowiedzi w Badaniu 1 i Badaniu 2. Podobieństwo pomiędzy XELODA 500 mg i 5-FU/LV w tych badaniach oceniano, analizując potencjalną różnicę między tymi dwoma rodzajami leczenia. Aby zapewnić, że XELODA 500 mg ma klinicznie istotny wpływ na przeżycie, przeprowadzono analizy statystyczne w celu określenia procentu wpływu 5-FU/LV na przeżycie, który został zachowany przez XELODA. Oszacowanie efektu przeżycia 5-FU/LV pochodziło z metaanalizy dziesięciu randomizowanych badań z opublikowanej literatury porównującej 5-FU ze schematami 5-FU/LV podobnymi do grup kontrolnych stosowanych w tych badaniach 1 i 2. Metodą porównania terapii było zbadanie najgorszego przypadku (95% górna granica ufności) pod kątem różnicy między 5-FU/LV a XELODA 500 mg i wykazanie, że utrata ponad 50% 5-FU/LV Wykluczono efekt przeżycia FU/LV. Wykazano, że odsetek utrzymania efektu 5-FU/LV na przeżycie wynosił co najmniej 61% w badaniu 2 i 10% w badaniu 1. Łączny wynik jest zgodny z zachowaniem co najmniej 50% efektu 5 -FU/LV. Należy zauważyć, że te wartości zachowania efektu są oparte na górnej granicy różnicy 5-FU/LV vs XELODA 500 mg. Wyniki te nie wykluczają możliwości prawdziwej równoważności produktu XELODA 500 mg z 5-FU/LV (patrz i Rysunek 3 ).
Rak piersi
XELODA została oceniona w badaniach klinicznych w połączeniu z docetakselem (Taxotere®) oraz w monoterapii.
W połączeniu z Docetakselem
Dawka XELODA zastosowana w badaniu klinicznym fazy 3 w skojarzeniu z docetakselem została oparta na wynikach badania fazy 1, w którym zakres dawek docetakselu podawanych w cyklach 3-tygodniowych w połączeniu z przerywanym schematem leczenia XELODA (14 dni) leczenia, po którym nastąpił 7-dniowy okres odpoczynku). Schemat dawkowania skojarzonego został wybrany na podstawie profilu tolerancji 75 mg/m2 podawanego w 3-tygodniowych cyklach docetakselu w połączeniu z 1250 mg/m2 dwa razy dziennie przez 14 dni XELODA 500 mg podawanego w 3-tygodniowych cyklach. Zatwierdzona dawka 100 mg/m2 docetakselu podawana w cyklach 3-tygodniowych była ramieniem kontrolnym badania III fazy.
XELODA w skojarzeniu z docetakselem oceniano w otwartym, wieloośrodkowym, randomizowanym badaniu w 75 ośrodkach w Europie, Ameryce Północnej, Ameryce Południowej, Azji i Australii. Do badania włączono łącznie 511 pacjentek z rakiem piersi z przerzutami, opornym na leczenie lub nawrotem w trakcie lub po leczeniu zawierającym antracykliny lub z nawrotem w trakcie lub nawrotem w ciągu 2 lat od zakończenia leczenia uzupełniającego zawierającego antracykliny. Dwieście pięćdziesięciu pięciu (255) pacjentów zostało losowo przydzielonych do grupy otrzymującej XELODA 1250 mg/m2 dwa razy na dobę przez 14 dni, a następnie 1 tydzień bez leczenia oraz docetaksel 75 mg/m2 w postaci 1-godzinnego wlewu dożylnego podawanego w 3-tygodniowych cyklach. W ramieniu monoterapii 256 pacjentów otrzymywało docetaksel w dawce 100 mg/m2 w postaci 1-godzinnego wlewu dożylnego w 3-tygodniowych cyklach. Dane demograficzne pacjentów przedstawiono w Tabeli 16.
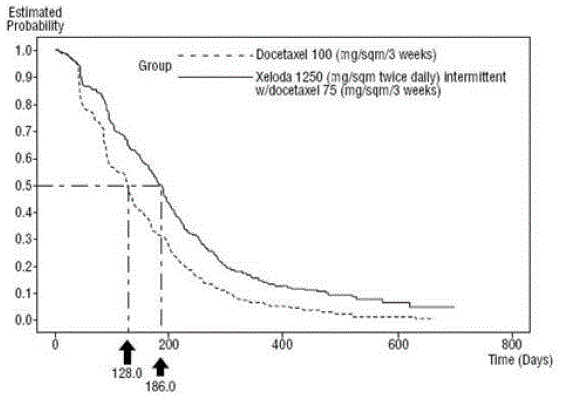
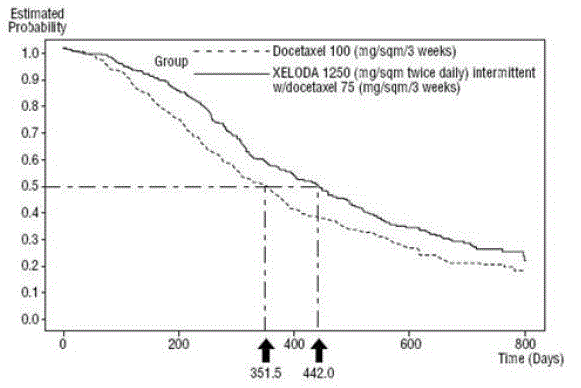
Produkt XELODA w skojarzeniu z docetakselem spowodował statystycznie istotną poprawę czasu do progresji choroby, całkowitego przeżycia i odsetka obiektywnych odpowiedzi w porównaniu z monoterapią docetakselem, jak pokazano w i Rysunek 5 .
Rycina 4 Szacunki Kaplana-Meiera dotyczące czasu do progresji choroby XELODA i Docetaksel vs Docetaksel
Rysunek 5 Szacunki Kaplana-Meiera dotyczące przeżycia XELODA i Docetaksel vs Docetaksel
Monoterapia
Działanie przeciwnowotworowe preparatu XELODA w monoterapii oceniano w otwartym, jednoramiennym badaniu przeprowadzonym w 24 ośrodkach w USA i Kanadzie. Do badania włączono 162 pacjentki z rakiem piersi w IV stopniu zaawansowania. Pierwszorzędowym punktem końcowym był odsetek odpowiedzi guza u pacjentów z mierzalną chorobą, z odpowiedzią zdefiniowaną jako ≥50% spadek sumy iloczynów prostopadłych średnic choroby mierzalnej dwuwymiarowo przez co najmniej 1 miesiąc. Produkt XELODA podawano w dawce 1255 mg/m2 dwa razy na dobę przez 2 tygodnie, po czym następował 1-tygodniowy okres odpoczynku i podawano w cyklach 3-tygodniowych. Wyjściowe dane demograficzne i charakterystykę kliniczną wszystkich pacjentów (n=162) oraz tych z mierzalną chorobą (n=135) przedstawiono w . Oporność zdefiniowano jako postępującą chorobę w trakcie leczenia, z początkową odpowiedzią lub bez, lub nawrót w ciągu 6 miesięcy od zakończenia leczenia uzupełniającym schematem chemioterapii zawierającym antracykliny.
Odpowiedzi przeciwnowotworowe u pacjentów z chorobą oporną zarówno na paklitaksel, jak i na antracyklinę przedstawiono w:
W podgrupie 43 pacjentów, którzy byli podwójnie oporni, mediana czasu do progresji wyniosła 102 dni, a mediana przeżycia 255 dni. Odsetek obiektywnych odpowiedzi w tej populacji został poparty wskaźnikiem odpowiedzi wynoszącym 18,5% (1 CR, 24 PR) w ogólnej populacji 135 pacjentów z mierzalną chorobą, którzy byli mniej oporni na chemioterapię (patrz ). Mediana czasu do progresji wyniosła 90 dni, a mediana przeżycia 306 dni.
INFORMACJA O PACJENCIE
XELODA® (zeh-LOE-duh) (kapecytabina) tabletki
Jakie są najważniejsze informacje, które powinienem wiedzieć o XELODA 500mg?
XELODA 500mg może powodować poważne skutki uboczne, w tym:
- XELODA może wchodzić w interakcje z lekami rozrzedzającymi krew, takimi jak warfaryna (COUMADIN®). Przyjmowanie leku XELODA 500 mg z tymi lekami może powodować zmiany w szybkości krzepnięcia krwi i może powodować krwawienie, które może prowadzić do śmierci. Może to nastąpić już kilka dni po rozpoczęciu przyjmowania leku XELODA 500 mg lub później w trakcie leczenia, a być może nawet w ciągu 1 miesiąca od zaprzestania przyjmowania leku XELODA. Twoje ryzyko może być wyższe, ponieważ masz raka i masz ponad 60 lat.
- Przed przyjęciem leku XELODA należy poinformować lekarza o przyjmowaniu warfaryny (COUMADIN) lub innego leku rozrzedzającego krew.
- Jeśli pacjent przyjmuje warfarynę (COUMADIN) lub inny lek rozrzedzający krew podobny do warfaryny (COUMADIN) podczas leczenia lekiem XELODA, lekarz prowadzący powinien często zlecać badania krwi, aby sprawdzić, jak szybko dochodzi do krzepnięcia krwi podczas i po zakończeniu leczenia lekiem XELODA. Twój lekarz może w razie potrzeby zmienić dawkę leku rozrzedzającego krew.
Widzieć „Jakie są możliwe skutki uboczne XELODA 500mg?” aby uzyskać więcej informacji na temat skutków ubocznych.
Co to jest XELODA?
XELODA to lek na receptę stosowany w leczeniu osób z:
- rak okrężnicy, który rozprzestrzenił się na węzły chłonne w okolicy okrężnicy (stadium Dukes C), po operacji.
- raka okrężnicy lub odbytnicy (jelita i odbytnicy), który rozprzestrzenił się na inne części ciała (przerzuty), jako pierwsze leczenie raka na tym etapie.
- raka piersi, który rozprzestrzenił się na inne części ciała (przerzuty) wraz z innym lekiem zwanym docetakselem, po tym, jak leczenie niektórymi innymi lekami przeciwnowotworowymi nie przyniosło skutku.
- raka piersi, który rozprzestrzenił się na inne części ciała i który nie uległ poprawie po leczeniu paklitakselem i niektórymi innymi lekami przeciwnowotworowymi, lub które nie mogą otrzymać dalszego leczenia niektórymi lekami przeciwnowotworowymi.
Nie wiadomo, czy XELODA 500mg jest bezpieczna i skuteczna u dzieci.
Nie stosować leku XELODA, jeśli:
- masz poważne problemy z nerkami
- jesteś uczulony na kapecytabinę, 5-fluorouracyl lub którykolwiek z jej składników zawartych w produkcie XELODA. Pełna lista składników leku XELODA znajduje się na końcu tej ulotki.
Przed przyjęciem leku XELODA należy porozmawiać z lekarzem, jeśli pacjent nie ma pewności, czy cierpi na którykolwiek z wyżej wymienionych schorzeń.
Przed przyjęciem leku XELODA należy poinformować lekarza o wszystkich swoich schorzeniach, w tym o:
Widzieć „Jakie są najważniejsze informacje, które powinienem wiedzieć o XELODA?”
- miałeś problemy z sercem
- masz problemy z nerkami lub wątrobą
- powiedziano Ci, że brakuje Ci enzymu DPD (dehydrogenazy dihydropirymidynowej).
- są w ciąży lub planują zajść w ciążę. XELODA 500 mg może zaszkodzić nienarodzonemu dziecku. Twój lekarz powinien wykonać test ciążowy przed rozpoczęciem leczenia lekiem XELODA. Należy natychmiast poinformować lekarza, jeśli pacjentka zajdzie w ciążę lub podejrzewa, że może być w ciąży podczas leczenia lekiem XELODA.
- Kobiety które mogą zajść w ciążę, powinny stosować skuteczną antykoncepcję podczas leczenia i przez 6 miesięcy po przyjęciu ostatniej dawki. Porozmawiaj z lekarzem o możliwościach kontroli urodzeń, które mogą być dla Ciebie odpowiednie podczas leczenia lekiem XELODA.
- Mężczyźni które mają partnerki zdolne do zajścia w ciążę, powinny stosować skuteczną kontrolę urodzeń w trakcie leczenia i przez 3 miesiące po przyjęciu ostatniej dawki.
- karmisz piersią lub planujesz karmić piersią. Nie wiadomo, czy XELODA przenika do mleka matki. Nie należy karmić piersią podczas leczenia lekiem XELODA 500 mg oraz przez 2 tygodnie po przyjęciu ostatniej dawki.
Poinformuj swojego lekarza o wszystkich przyjmowanych lekach, w tym o lekach na receptę i dostępnych bez recepty, witaminach i suplementach ziołowych. XELODA może wpływać na działanie innych leków, a inne leki mogą wpływać na działanie leku XELODA.
Poznaj leki, które przyjmujesz. Zachowaj ich listę, aby pokazać swojemu lekarzowi i farmaceucie, kiedy otrzymasz nowy lek.
Jak zażywać XELODA 500mg?
- XELODA należy przyjmować dokładnie tak, jak zalecił jej lekarz.
- Twój lekarz poinformuje Cię, jaką dawkę leku XELODA należy przyjąć i kiedy.
- XELODA należy przyjmować 2 razy dziennie, 1 raz rano i 1 raz wieczorem.
- Przyjmuj XELODA 500mg w ciągu 30 minut po zakończeniu posiłku.
- Tabletki XELODA 500 mg należy połknąć w całości, popijając wodą. Nie rób zmiażdżyć lub pokroić tabletki XELODA 500mg. Jeśli pacjent nie może połknąć tabletek XELODA w całości, należy poinformować o tym lekarza.
- Twój lekarz może zmienić dawkę, tymczasowo lub na stałe przerwać leczenie lekiem XELODA 500 mg, jeśli wystąpią działania niepożądane.
- Jeśli zażyjesz zbyt dużą dawkę leku XELODA, natychmiast skontaktuj się z lekarzem lub udaj się na pogotowie ratunkowe najbliższego szpitala.
Jakie są możliwe skutki uboczne XELODA?
XELODA może powodować poważne skutki uboczne, w tym:
Nudności i wymioty są powszechne w przypadku produktu XELODA. Jeśli stracisz apetyt, poczujesz się słabo, masz nudności, wymioty lub biegunkę, możesz szybko się odwodnić.
Przestań przyjmować lek XELODA i natychmiast skontaktuj się z lekarzem, jeśli:
Jeśli liczba białych krwinek jest bardzo niska, istnieje zwiększone ryzyko infekcji. Zadzwoń do swojego lekarza natychmiast, jeśli wystąpi gorączka 100, 5oF lub wyższa lub wystąpią inne oznaki i objawy infekcji.
- Widzieć „Jakie są najważniejsze informacje, które powinienem wiedzieć o XELODA?”
- Biegunka. Biegunka jest powszechna w przypadku leku XELODA i czasami może być ciężka. Należy przerwać przyjmowanie leku XELODA i natychmiast skontaktować się z lekarzem, jeśli liczba wypróżnień w ciągu dnia zwiększy się o 4 lub więcej wypróżnień niż zwykle lub w przypadku wypróżnień w nocy. Zapytaj swojego lekarza o leki, które możesz zażywać w celu leczenia biegunki. Jeśli masz ciężką krwawą biegunkę z silnym bólem brzucha i gorączką, przerwij przyjmowanie leku Xeloda 500 mg i natychmiast skontaktuj się z lekarzem lub udaj się na pogotowie ratunkowe najbliższego szpitala.
- Problemy sercowe. XELODA może powodować problemy z sercem, w tym: zawał serca i zmniejszony przepływ krwi do serca, ból w klatce piersiowej, nieregularne bicie serca, zmiany w aktywności elektrycznej serca widoczne w elektrokardiogramie (EKG), problemy z mięśniem sercowym, niewydolność serca i nagłe śmierć. Przestań przyjmować lek XELODA 500 mg i natychmiast skontaktuj się z lekarzem lub udaj się na pogotowie ratunkowe najbliższego szpitala, jeśli wystąpią jakiekolwiek nowe objawy problemów z sercem, w tym:
- ból w klatce piersiowej
- zawroty głowy
- duszność
- zawroty
- Utrata zbyt dużej ilości płynów ustrojowych (odwodnienie) i niewydolność nerek. Odwodnienie może wystąpić podczas stosowania leku XELODA 500 mg i może spowodować nagłą niewydolność nerek, która może prowadzić do śmierci. Osoby, które przed przyjęciem leku XELODA 500 mg mają problemy z nerkami, a także przyjmują inne leki, które mogą powodować problemy z nerkami, są w grupie podwyższonego ryzyka.
- wymiotować 2 lub więcej razy dziennie.
- są w stanie tylko trochę jeść lub pić od czasu do czasu lub wcale z powodu nudności.
- mieć biegunkę. Patrz „biegunka” powyżej.
- Ciężkie reakcje skórne i ustne.
- XELODA 500mg może powodować ciężkie reakcje skórne, które mogą prowadzić do śmierci. Poinformuj natychmiast swojego lekarza, jeśli pojawi się wysypka skórna, pęcherze i łuszczenie się skóry. Lekarz może zalecić przerwanie stosowania leku XELODA w przypadku wystąpienia ciężkiej reakcji skórnej. W takim przypadku nie należy ponownie przyjmować leku XELODA 500 mg.
- XELODA 500mg może również powodować syndrom „ręki i stopy”. Zespół dłoni i stóp jest powszechny w przypadku leku XELODA 500 mg i może powodować drętwienie i zmiany czucia w dłoniach i stopach lub powodować zaczerwienienie, ból, obrzęk dłoni i stóp. Należy przerwać przyjmowanie leku XELODA i natychmiast skontaktować się z lekarzem, jeśli wystąpi którykolwiek z tych objawów i nie można wykonywać zwykłych czynności.
- Zespół dłoni i stóp może prowadzić do utraty odcisków palców, co może mieć wpływ na Twoją identyfikację.
- podczas przyjmowania leku XELODA mogą wystąpić owrzodzenia w jamie ustnej lub na języku. Należy przerwać przyjmowanie leku XELODA 500 mg i skontaktować się z lekarzem w przypadku bolesnego zaczerwienienia, obrzęku lub owrzodzenia jamy ustnej lub języka albo w przypadku problemów z jedzeniem.
- Zwiększony poziom bilirubiny we krwi i problemy z wątrobą. Podwyższona bilirubina we krwi jest powszechna w przypadku leku XELODA 500 mg i czasami może być również ciężka. Twój lekarz sprawdzi, czy nie występują te problemy podczas leczenia lekiem XELODA.
- Zmniejszona liczba białych krwinek, płytek krwi i czerwonych krwinek. Podczas leczenia lekiem XELODA lekarz wykona badania krwi w celu sprawdzenia liczby krwinek.
Osoby w wieku 80 lat lub starsze mogą być bardziej narażone na wystąpienie ciężkich lub poważnych działań niepożądanych związanych ze stosowaniem leku XELODA.
Najczęstsze skutki uboczne XELODA 500mg to:
- biegunka
- ból brzucha (brzucha)
- zespół dłoni i stóp
- osłabienie i zmęczenie
- mdłości
- zwymiotował
- zwiększone ilości produktów rozpadu krwinek czerwonych (bilirubina we krwi)
Po zastosowaniu leku XELODA mogą wystąpić ciężkie reakcje alergiczne. Poinformuj swojego lekarza, jeśli kiedykolwiek miałeś reakcję alergiczną na kapecytabinę lub 5-fluorouracyl. Widzieć „Nie należy przyjmować leku XELODA, jeśli:”. Przestań przyjmować lek XELODA 500 mg i natychmiast skontaktuj się z lekarzem lub udaj się na pogotowie, jeśli wystąpi którykolwiek z następujących objawów ciężkiej reakcji alergicznej:
- czerwone swędzące pręgi na skórze (pokrzywka)
- zaczerwienienie skóry
- obrzęk twarzy, ust, języka lub gardła
- wysypka
- swędzący
- kłopoty z połykaniem lub oddychaniem
XELODA 500mg może powodować problemy z płodnością u kobiet i mężczyzn. Może to wpłynąć na zdolność do posiadania dziecka. Porozmawiaj ze swoim lekarzem, jeśli masz obawy dotyczące płodności.
To nie wszystkie możliwe skutki uboczne XELODA.
Zadzwoń do lekarza, aby uzyskać poradę medyczną na temat skutków ubocznych. Możesz zgłosić skutki uboczne do FDA pod numerem 1-800-FDA-1088.
Jak przechowywać XELODĘ?
- Przechowywać XELODA 500mg w temperaturze pokojowej między 68 ° F do 77 ° F (20 ° C do 25 ° C).
- Przechowywać XELODA 500mg w szczelnie zamkniętym pojemniku.
- Należy zapytać lekarza lub farmaceutę, jak bezpiecznie wyrzucić niewykorzystany lek XELODA.
Lek XELODA i wszystkie leki należy przechowywać w miejscu niedostępnym dla dzieci.
Ogólne informacje o bezpiecznym i efektywnym korzystaniu z XELODA.
Czasami leki są przepisywane do celów innych niż wymienione w ulotce informacyjnej dla pacjenta. Nie należy stosować leku XELODA w stanach, na które nie został przepisany. Nie należy podawać leku XELODA 500mg innym osobom, nawet jeśli mają takie same objawy jak Ty. Może im to zaszkodzić. Możesz poprosić farmaceutę lub pracownika służby zdrowia o informacje na temat leku XELODA 500 mg, które są przeznaczone dla pracowników służby zdrowia.
Jakie są składniki XELODA 500mg?
Składnik czynny: kapecytabina
Nieaktywne składniki: laktoza bezwodna, kroskarmeloza sodowa, hydroksypropylometyloceluloza, celuloza mikrokrystaliczna, stearynian magnezu i woda oczyszczona. Powłoka brzoskwiniowa lub jasna brzoskwinia zawiera hydroksypropylometylocelulozę, talk, dwutlenek tytanu oraz syntetyczny żółty i czerwony tlenki żelaza.
Aby uzyskać więcej informacji, wejdź na http://www.gene.com/patients/medicines/xeloda 500mg lub zadzwoń pod numer 1-877-436-3683.
Niniejsza informacja dla pacjenta została zatwierdzona przez amerykańską Agencję ds. Żywności i Leków.