Rebetol 200mg Ribavirin Zastosowanie, skutki uboczne i dawkowanie. Cena w aptece internetowej. Leki generyczne bez recepty.
Co to jest Rebetol 200mg i jak się go stosuje?
Rebetol jest lekiem na receptę stosowanym w leczeniu objawów przewlekłego zapalenia wątroby typu C. Rebetol 200 mg może być stosowany samodzielnie lub z innymi lekami.
Rebetol należy do klasy leków zwanych wirusami zapalenia wątroby typu B/zapalenia wątroby typu C; Agenci RSV.
Nie wiadomo, czy Rebetol 200 mg jest bezpieczny i skuteczny u dzieci w wieku poniżej 3 lat.
Jakie są możliwe skutki uboczne Rebetolu?
Rebetol może powodować poważne działania niepożądane, w tym:
- pokrzywka,
- trudności w oddychaniu,
- obrzęk twarzy, warg, języka lub gardła,
- Problemy ze wzrokiem
- silny ból w górnej części brzucha rozprzestrzeniający się na plecy,
- mdłości,
- wymioty,
- biegunka,
- nowy lub nasilający się kaszel,
- gorączka,
- przeszywający ból w klatce piersiowej,
- świszczący oddech,
- duszność,
- ciężka depresja,
- myśli o samookaleczeniu,
- myśli o krzywdzeniu innych,
- blada lub pożółkła skóra,
- ciemny kolor moczu,
- dezorientacja,
- słabość,
- dreszcze,
- objawy grypopodobne,
- obrzęk dziąseł,
- owrzodzenia jamy ustnej,
- owrzodzenia skóry,
- łatwe siniaki,
- nietypowe krwawienie i
- zawroty
- Natychmiast uzyskaj pomoc medyczną, jeśli wystąpi którykolwiek z wymienionych powyżej objawów.
- Najczęstsze skutki uboczne Rebetolu to:
- mdłości,
- wymioty,
- utrata apetytu,
- gorączka,
- dreszcze,
- drżący,
- niska liczba krwinek,
- niedokrwistość,
- słabość,
- zmęczenie,
- ból głowy,
- ból w mięśniach,
- zmiany nastroju,
- niepokój i
- drażliwość
Poinformuj lekarza, jeśli wystąpią jakiekolwiek skutki uboczne, które Ci przeszkadzają lub które nie ustępują.
To nie wszystkie możliwe skutki uboczne Rebetolu. Aby uzyskać więcej informacji, skontaktuj się z lekarzem lub farmaceutą.
Zadzwoń do lekarza, aby uzyskać poradę medyczną na temat skutków ubocznych. Możesz zgłosić skutki uboczne do FDA pod numerem 1-800-FDA-1088.
OSTRZEŻENIE
RYZYKO POWAŻNYCH ZABURZEŃ I SKUTKÓW ZWIĄZANYCH Z RYBAWIRYNĄ
- Monoterapia REBETOL nie jest skuteczna w leczeniu przewlekłego zakażenia wirusem zapalenia wątroby typu C i nie powinna być stosowana w tym wskazaniu samodzielnie [patrz OSTRZEŻENIA I ŚRODKI OSTROŻNOŚCI].
- Podstawową toksycznością rybawiryny jest niedokrwistość hemolityczna. Niedokrwistość związana z terapią REBETOL 200 mg może prowadzić do pogorszenia choroby serca, która doprowadziła do zawału mięśnia sercowego prowadzącego do zgonu i niezakończonego zgonem. Pacjenci z historią poważnej lub niestabilnej choroby serca nie powinni być leczeni preparatem REBETOL [patrz DAWKOWANIE I PODAWANIE, OSTRZEŻENIA I ŚRODKI OSTROŻNOŚCI oraz DZIAŁANIA NIEPOŻĄDANE].
- wszystkich gatunków zwierząt narażonych na rybawirynę wykazano istotne działanie teratogenne i embriobójcze. Ponadto rybawiryna ma okres półtrwania po podaniu wielokrotnej dawki 12 dni, a zatem może utrzymywać się w przedziałach pozaosoczowych nawet przez 6 miesięcy. Dlatego terapia REBETOL jest przeciwwskazana u kobiet w ciąży oraz u partnerów kobiet w ciąży. Należy zachować szczególną ostrożność, aby uniknąć ciąży w trakcie leczenia i przez 6 miesięcy po zakończeniu leczenia zarówno u pacjentek, jak i u partnerek pacjentów płci męskiej przyjmujących preparat REBETOL. Podczas leczenia oraz w okresie 6 miesięcy po zakończeniu leczenia należy stosować co najmniej dwie niezawodne formy skutecznej antykoncepcji [patrz PRZECIWWSKAZANIA, OSTRZEŻENIA I ŚRODKI OSTROŻNOŚCI, Stosowanie w określonych populacjach, Toksykologia niekliniczna oraz INFORMACJE DLA PACJENTA].
OPIS
REBETOL (rybawiryna) to syntetyczny analog nukleozydowy (analog puryny). Nazwa chemiczna rybawiryny to 1-β-D-rybofuranozylo-1H-1,2,4-triazolo-3-karboksyamid i ma następujący wzór strukturalny (patrz Rysunek 1).
Rysunek 1: Wzór strukturalny
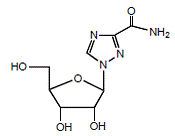
Rybawiryna jest białym, krystalicznym proszkiem. Jest dobrze rozpuszczalny w wodzie i słabo rozpuszczalny w bezwodnym alkoholu. Wzór empiryczny to C8H12N4O5, a masa cząsteczkowa to 244,21.
Kapsułki REBETOL składają się z białego proszku w białej, nieprzezroczystej, żelatynowej kapsułce. Każda kapsułka zawiera 200 mg rybawiryny oraz nieaktywne składniki: celulozę mikrokrystaliczną, monohydrat laktozy, kroskarmelozę sodową i stearynian magnezu. Otoczka kapsułki składa się z żelatyny, laurylosiarczanu sodu, dwutlenku krzemu i dwutlenku tytanu. Kapsułka jest nadrukowana jadalnym niebieskim tuszem farmaceutycznym, który składa się z szelaku, bezwodnego alkoholu etylowego, alkoholu izopropylowego, alkoholu n-butylowego, glikolu propylenowego, wodorotlenku amonu i lak aluminiowy FD&C Blue #2.
REBETOL 200 mg roztwór doustny jest przezroczystym, bezbarwnym lub jasnożółtym płynem o smaku gumy balonowej. Każdy mililitr roztworu zawiera 40 mg rybawiryny i nieaktywne składniki: sacharozę, glicerynę, sorbitol, glikol propylenowy, cytrynian sodu, kwas cytrynowy, benzoesan sodu, naturalny i sztuczny aromat do gumy balonowej #15864 oraz wodę.
WSKAZANIA
Przewlekłe wirusowe zapalenie wątroby typu C (CHC)
REBETOL® (rybawiryna) w połączeniu z interferonem alfa-2b (pegylowanym i niepegylowanym) jest wskazany w leczeniu przewlekłego zapalenia wątroby typu C (CHC) u pacjentów w wieku 3 lat i starszych z wyrównaną chorobą wątroby [patrz OSTRZEŻENIA I ŚRODKI , oraz Stosowanie w określonych populacjach ].
Rozpoczynając terapię skojarzoną REBETOL z PegIntron® lub INTRON A® należy wziąć pod uwagę następujące punkty:
- Wskazania te opierają się na osiągnięciu niewykrywalnego HCV-RNA po leczeniu przez 24 lub 48 tygodni i utrzymaniu trwałej odpowiedzi wirusologicznej (SVR) 24 tygodnie po podaniu ostatniej dawki.
- Terapia skojarzona z REBETOL/PegIntron jest preferowana w porównaniu z REBETOL/INTRON A, ponieważ ta kombinacja zapewnia znacznie lepsze wskaźniki odpowiedzi [patrz Studia kliniczne ].
- Pacjenci z następującymi cechami mają mniejsze szanse na odniesienie korzyści z ponownego leczenia po niepowodzeniu leczenia: poprzedni brak odpowiedzi, wcześniejsze leczenie pegylowanym interferonem, znaczące zwłóknienie pomostowe lub marskość wątroby oraz zakażenie o genotypie 1 [patrz Studia kliniczne ].
- Brak danych dotyczących bezpieczeństwa i skuteczności leczenia dłuższego niż jeden rok.
DAWKOWANIE I SPOSÓB PODAWANIA
Pod żadnym pozorem nie wolno otwierać, kruszyć ani łamać kapsułek REBETOL. REBETOL 200 mg należy przyjmować z jedzeniem [patrz FARMAKOLOGIA KLINICZNA ]. REBETOL nie powinien być stosowany u pacjentów z klirensem kreatyniny mniejszym niż 50 ml/min.
Terapia skojarzona REBETOL/PegIntron
Pacjenci dorośli
Zalecana dawka preparatu PegIntron wynosi 1,5 µg/kg/tydzień podskórnie w połączeniu z 800 do 1400 mg kapsułki REBETOL 200 mg doustnie, w zależności od masy ciała pacjenta (patrz Tabela 1). Objętość wstrzykiwanego produktu PegIntron zależy od siły produktu PegIntron i masy ciała pacjenta. Dodatkowe informacje dotyczące dawkowania znajdują się na etykiecie produktu PegIntron.
Czas trwania leczenia – interferon alfa-naiwni pacjenci
Czas leczenia pacjentów z genotypem 1 wynosi 48 tygodni. Należy rozważyć przerwanie leczenia u pacjentów, którzy nie osiągnęli co najmniej 2 log10 spadku lub utraty HCV-RNA po 12 tygodniach lub jeśli HCV-RNA pozostaje wykrywalne po 24 tygodniach leczenia. Pacjenci z genotypem 2 i 3 powinni być leczeni przez 24 tygodnie.
Czas trwania leczenia – Ponowne leczenie za pomocą PegIntron/REBETOL w przypadku niepowodzeń wcześniejszego leczenia
Czas leczenia dla pacjentów, u których wcześniej nie powiodła się terapia, wynosi 48 tygodni, niezależnie od genotypu HCV. Pacjenci ponownie leczeni, u których nie uzyskano niewykrywalnego HCV-RNA w 12. tygodniu terapii lub u których HCV-RNA pozostaje wykrywalny po 24 tygodniach leczenia, jest bardzo mało prawdopodobne, aby osiągnęli SVR i należy rozważyć przerwanie leczenia [patrz Studia kliniczne ].
Pacjenci pediatryczni
Dawkowanie u pacjentów pediatrycznych określa się na podstawie powierzchni ciała w przypadku preparatu PegIntron oraz masy ciała w przypadku preparatu REBETOL. Zalecana dawka preparatu PegIntron wynosi 60 μg/m²/tydzień podskórnie w skojarzeniu z 15 mg/kg/dobę preparatu REBETOL 200 mg doustnie w dwóch dawkach podzielonych (patrz Tabela 2) u dzieci w wieku 3-17 lat. Pacjenci, którzy osiągną 18. rok życia podczas otrzymywania leku PegIntron/REBETOL 200 mg, powinni nadal stosować pediatryczny schemat dawkowania. Czas leczenia pacjentów z genotypem 1 wynosi 48 tygodni. Pacjenci z genotypem 2 i 3 powinni być leczeni przez 24 tygodnie.
Terapia skojarzona REBETOL/INTRON A
Dorośli ludzie
Czas trwania leczenia – interferon alfa-naiwni pacjenci
Zalecana dawka preparatu INTRON A wynosi 3 miliony jm trzy razy w tygodniu podskórnie. Zalecana dawka kapsułek REBETOL zależy od masy ciała pacjenta (patrz Tabela 3). Zalecany czas trwania leczenia u pacjentów wcześniej nieleczonych interferonem wynosi od 24 do 48 tygodni. Czas trwania leczenia należy dostosować indywidualnie do pacjenta w zależności od wyjściowej charakterystyki choroby, odpowiedzi na leczenie i tolerancji schematu [patrz WSKAZANIA I ZASTOSOWANIE , DZIAŁANIA NIEPOŻĄDANE , oraz Studia kliniczne ]. Po 24 tygodniach leczenia należy ocenić odpowiedź wirusologiczną. Należy rozważyć przerwanie leczenia u każdego pacjenta, u którego w ciągu 24 tygodni nie uzyskano HCV-RNA poniżej granicy wykrywalności testu. Brak danych dotyczących bezpieczeństwa i skuteczności leczenia trwającego dłużej niż 48 tygodni w populacji pacjentów wcześniej nieleczonych.
Czas trwania leczenia – Ponowne leczenie preparatem INTRON A/REBETOL u pacjentów z nawrotami
pacjentów z nawrotem po monoterapii niepegylowanym interferonem zalecany czas leczenia wynosi 24 tygodnie.
Pediatria
Zalecana dawka preparatu REBETOL to 15 mg/kg na dobę doustnie (dawka podzielona rano i wieczorem). Patrz Tabela 2 dla dawkowania produktu REBETOL u dzieci w skojarzeniu z INTRON A. INTRON A do wstrzykiwań na masę ciała od 25 kg do 61 kg wynosi 3 miliony j.m./m2 podskórnie trzy razy w tygodniu. W przypadku masy ciała większej niż 61 kg należy zapoznać się z tabelą dawkowania dla dorosłych.
Zalecany czas trwania leczenia wynosi 48 tygodni u dzieci i młodzieży z genotypem 1. Po 24 tygodniach leczenia należy ocenić odpowiedź wirusologiczną. Należy rozważyć przerwanie leczenia u każdego pacjenta, który do tego czasu nie osiągnął HCV-RNA poniżej granicy wykrywalności testu. Zalecany czas trwania leczenia u dzieci z genotypem 2/3 wynosi 24 tygodnie.
Testy laboratoryjne
Poniższe badania laboratoryjne są zalecane u wszystkich pacjentów leczonych preparatem REBETOL 200 mg przed rozpoczęciem leczenia, a następnie okresowo.
Standardowe testy hematologiczne – w tym hemoglobina (wstępne leczenie, tydzień 2 i tydzień 4 terapii, a także w przypadkach uzasadnionych klinicznie [patrz OSTRZEŻENIA I ŚRODKI ], całkowitą i różnicową liczbę białych krwinek oraz liczbę płytek krwi.
- Chemia krwi - testy czynności wątroby i TSH.
- Ciąża - w tym comiesięczny monitoring kobiet w wieku rozrodczym.
- EKG [patrz OSTRZEŻENIA I ŚRODKI ].
Modyfikacje dawki
Jeśli podczas leczenia skojarzonego REBETOL/INTRON A lub REBETOL/PegIntron wystąpią ciężkie działania niepożądane lub nieprawidłowe wyniki badań laboratoryjnych, należy zmodyfikować dawkę lub przerwać dawkowanie do czasu ustąpienia lub zmniejszenia nasilenia działania niepożądanego [patrz OSTRZEŻENIA I ŚRODKI ]. Jeśli nietolerancja utrzymuje się po dostosowaniu dawki, należy przerwać leczenie skojarzone. Zmniejszenie dawki preparatu PegIntron u dorosłych pacjentów stosujących skojarzoną terapię REBETOL/PegIntron jest realizowane w dwuetapowym procesie od początkowej dawki początkowej 1,5 µg/kg/tydzień, do 1 µg/kg/tydzień, a następnie do 0,5 µg/kg/tydzień , Jeśli potrzebne. Dodatkowe informacje dotyczące zmniejszania dawki produktu PegIntron można znaleźć na etykiecie produktu PegIntron.
W badaniu 2 dotyczącym terapii skojarzonej u dorosłych zmniejszenie dawki wystąpiło u 42% pacjentów otrzymujących PegIntron 1,5 µg/kg i REBETOL 800 mg na dobę, w tym 57% osób o masie ciała 60 kg lub mniejszej. W badaniu 4 u 16% pacjentów zmniejszono dawkę PegIntronu do 1 µg/kg w skojarzeniu z REBETOLEM, a dodatkowe 4% wymagało drugiego zmniejszenia dawki PegIntronu do 0,5 µg/kg z powodu działań niepożądanych [patrz DZIAŁANIA NIEPOŻĄDANE ].
Zmniejszenie dawki u pacjentów pediatrycznych osiąga się poprzez dwuetapową modyfikację zalecanej dawki produktu PegIntron z pierwotnej dawki początkowej 60 μg/m²/tydzień, do 40 μg/m²/tydzień, a następnie do 20 μg/m²/tydzień, jeśli potrzebne (patrz Tabela 4). W badaniu pediatrycznym leczenia skojarzonego zmniejszenie dawki wystąpiło u 25% pacjentów otrzymujących PegIntron 60 μg/m2 raz na tydzień i REBETOL 15 mg/kg raz na dobę. Zmniejszenie dawki u dzieci osiąga się poprzez zmianę zalecanej dawki REBETOL z pierwotnej dawki początkowej 15 mg/kg na dobę w procesie dwuetapowym do 12 mg/kg/dobę, a następnie, w razie potrzeby, do 8 mg/kg/dobę ( patrz Tabela 4).
REBETOL 200mg nie powinien być stosowany u pacjentów z klirensem kreatyniny mniejszym niż 50 ml/min. Pacjenci z zaburzeniami czynności nerek i osoby w wieku powyżej 50 lat powinni być uważnie obserwowani pod kątem rozwoju niedokrwistości [patrz OSTRZEŻENIA I ŚRODKI , Używaj w określonych populacjach , oraz FARMAKOLOGIA KLINICZNA ].
REBETOL 200 mg należy podawać ostrożnie pacjentom z istniejącą wcześniej chorobą serca. Pacjentów należy ocenić przed rozpoczęciem leczenia i odpowiednio monitorować w trakcie leczenia. W przypadku jakiegokolwiek pogorszenia stanu sercowo-naczyniowego leczenie należy przerwać [patrz OSTRZEŻENIA I ŚRODKI ].
U pacjentów ze stabilną chorobą sercowo-naczyniową w wywiadzie wymagane jest trwałe zmniejszenie dawki, jeśli stężenie hemoglobiny zmniejszy się o więcej lub równo 2 g/dl w dowolnym okresie 4 tygodni. Ponadto, w przypadku tych pacjentów w wywiadzie kardiologicznym, jeśli stężenie hemoglobiny pozostaje poniżej 12 g/dl po 4 tygodniach przy zmniejszonej dawce, pacjent powinien przerwać terapię skojarzoną.
Zaleca się, aby u pacjenta, u którego poziom hemoglobiny spadł poniżej 10 g/dl, zmodyfikowano dawkę produktu REBETOL lub odstawiono go zgodnie z Tabelą 4 [patrz OSTRZEŻENIA I ŚRODKI ].
Dodatkowe informacje na temat zmniejszania dawki INTRON A lub PegIntron można znaleźć na etykietach INTRON A lub PegIntron.
Przerwanie dawkowania
Dorośli ludzie
pacjentów z genotypem 1 HCV, uprzednio nieleczonych interferonem, otrzymujących PegIntron w skojarzeniu z rybawiryną, zaleca się przerwanie leczenia, jeśli nie nastąpi spadek lub utrata HCV-RNA o co najmniej 2 log10 w ciągu 12 tygodni leczenia lub jeśli HCV-RNA poziomy pozostają wykrywalne po 24 tygodniach terapii. Niezależnie od genotypu, prawdopodobieństwo osiągnięcia SVR u wcześniej leczonych pacjentów z wykrywalnym HCV-RNA w 12. lub 24. tygodniu jest bardzo mało prawdopodobne i należy rozważyć przerwanie leczenia.
Pediatria (3-17 lat)
Zaleca się, aby pacjenci otrzymujący połączenie PegIntron/REBETOL (z wyjątkiem HCV Genotyp 2 i 3) przerwali leczenie po 12 tygodniach, jeśli ich RNA HCV w 12. tygodniu leczenia spadło o mniej niż 2 log10 w porównaniu z HCV-RNA w 24. tygodniu leczenia.
JAK DOSTARCZONE
Formy dawkowania i mocne strony
REBETOL 200 mg kapsułki 200 mg
REBETOL 200 mg roztwór doustny 40 mg na ml
Składowania i stosowania
REBETOL 200 mg kapsułki są białymi, nieprzezroczystymi kapsułkami z REBETOL, 200 mg i logo Schering Corporation nadrukowanym na otoczce kapsułki; kapsułki pakowane są w butelkę zawierającą 56 kapsułek ( NDC 0085-1351-05), 70 kapsułek ( NDC 0085-1385-07) i 84 kapsułki ( NDC 0085-1194-03).
REBETOL 200 mg roztwór doustny 40 mg na ml jest przezroczystym, bezbarwnym lub jasnożółtym płynem o smaku gumy balonowej i jest pakowany w 4-uncjowe butelki ze szkła bursztynowego (100 ml/butelkę) z zamknięciami zabezpieczającymi przed dostępem dzieci ( NDC 0085-1318-01).
Butelkę z kapsułkami REBETOL należy przechowywać w temperaturze 25°C (77°F); wycieczki dozwolone do 15-30°C (59-86°F) [patrz Temperatura pokojowa kontrolowana przez USP ].
REBETOL 200 mg roztwór doustny należy przechowywać w temperaturze 2-8°C (36-46°F) lub 25°C (77°F); wycieczki dozwolone do 15-30°C (59-86°F) [patrz Temperatura pokojowa kontrolowana przez USP ].
REBETOL 200 mg roztwór doustny wyprodukowany dla: Merck Sharp & Dohme Corp., filii Whitehouse Station, NJ 08889, USA. Producent: Schering-Plough Canada, Inc., Pointe Claire, Quebec, Kanada Kapsułki REBETOL produkowane przez: Merck Sharp & Dohme Corp., spółkę zależną Whitehouse Station, NJ 08889, USA. Aktualizacja: grudzień 2014 r.
SKUTKI UBOCZNE
Badania kliniczne preparatu REBETOL w skojarzeniu z PegIntron lub INTRON A przeprowadzono na ponad 7800 osobach w wieku od 3 do 76 lat.
Podstawową toksycznością rybawiryny jest niedokrwistość hemolityczna. Obniżenie poziomu hemoglobiny wystąpiło w ciągu pierwszych 1-2 tygodni terapii doustnej. Reakcje serca i płuc związane z niedokrwistością wystąpiły u około 10% pacjentów [patrz OSTRZEŻENIA I ŚRODKI ].
ponad 96% wszystkich uczestników badań klinicznych wystąpiło jedno lub więcej działań niepożądanych. Najczęściej zgłaszanymi działaniami niepożądanymi u dorosłych pacjentów otrzymujących PegIntron lub INTRON A w skojarzeniu z REBETOL 200 mg były zapalenie/reakcja w miejscu wstrzyknięcia, zmęczenie/astenia, ból głowy, dreszcze, gorączka, nudności, bóle mięśni i niepokój/chwiejność emocjonalna/drażliwość. Najczęstszymi działaniami niepożądanymi u dzieci w wieku 3 lat i starszych otrzymujących REBETOL 200 mg w skojarzeniu z PegIntronem lub INTRON A były gorączka, ból głowy, neutropenia, zmęczenie, jadłowstręt, rumień w miejscu wstrzyknięcia i wymioty.
Sekcja „Działania niepożądane” odnosi się do następujących badań klinicznych:
- Badania terapii skojarzonej REBETOL/PegIntron:
- Badanie kliniczne 1 – oceniane monoterapię PegIntronem (nieopisane dalej na tej etykiecie; informacje na temat tego badania znajdują się na etykiecie produktu PegIntron).
- Badanie 2 – oceniano REBETOL 800 mg/dobę stałą dawkę w połączeniu z 1,5 mcg/kg/tydzień PegIntron lub z INTRON A.
- Badanie 3 – oceniano PegIntron/w oparciu o masę ciała REBETOL 200 mg w skojarzeniu z PegIntronem/stałą dawką REBETOL 200 mg.
- Badanie 4 – porównano dwie dawki PegIntronu (1,5 μg/kg/tydzień i 1 μg/kg/tydzień) w skojarzeniu z REBETOLem i trzecią grupą leczoną otrzymującą Pegasys® (180 μg/tydzień)/Copegus® (1000-1200 mg/dzień ).
- Badanie 5 – oceniano PegIntron (1,5 µg/kg/tydzień) w skojarzeniu z preparatem REBETOL opartym na masie ciała u pacjentów, u których wcześniejsze leczenie zakończyło się niepowodzeniem.
- PegIntron/REBETOL 200 mg terapia skojarzona u pacjentów pediatrycznych
- Badania REBETOL/INTRON A w terapii skojarzonej dla dorosłych i dzieci
Poważne działania niepożądane wystąpiły u około 12% pacjentów w badaniach klinicznych produktu PegIntron z preparatem REBETOL lub bez REBETOL [patrz OSTRZEŻENIE W PUDEŁKU , OSTRZEŻENIA I ŚRODKI ]. Najczęstszymi poważnymi zdarzeniami występującymi u osób leczonych preparatami PegIntron i REBETOL 200 mg były depresja i myśli samobójcze [patrz OSTRZEŻENIA I ŚRODKI ], z których każdy występuje z częstotliwością mniejszą niż 1%. Myśli lub próby samobójcze występowały częściej u pacjentów pediatrycznych, głównie młodzieży, w porównaniu z pacjentami dorosłymi (2,4% w porównaniu z 1%) w trakcie leczenia i obserwacji po zakończeniu terapii [patrz OSTRZEŻENIA I ŚRODKI ]. Najczęstszą reakcją śmiertelną występującą u osób leczonych preparatami PegIntron i REBETOL 200 mg było zatrzymanie akcji serca, myśli samobójcze i próby samobójcze [patrz OSTRZEŻENIA I ŚRODKI ], wszystkie występujące u mniej niż 1% badanych.
Ponieważ badania kliniczne prowadzone są w bardzo zróżnicowanych warunkach, częstość występowania działań niepożądanych obserwowanych w badaniach klinicznych leku nie może być bezpośrednio porównywana z częstością w badaniach klinicznych innego leku i może nie odzwierciedlać częstości obserwowanych w praktyce klinicznej.
Doświadczenie w badaniach klinicznych – Terapia skojarzona REBETOL/PegIntron
Osoby dorosłe
Działania niepożądane, które wystąpiły w badaniu klinicznym z częstością większą niż 5%, przedstawiono w Tabeli 5 według grup leczonych z terapii skojarzonej REBETOL/PegIntron (badanie 2).
W Tabeli 6 podsumowano związane z leczeniem działania niepożądane w Badaniu 4, które wystąpiły z częstością większą lub równą 10%.
Częstość występowania ciężkich działań niepożądanych była porównywalna we wszystkich badaniach. W badaniu 3 zaobserwowano podobną częstość występowania ciężkich działań niepożądanych zgłoszonych w grupie stosującej REBETOL w oparciu o masę ciała (12%) i w schemacie z płaską dawką REBETOL. W badaniu 2 częstość występowania ciężkich działań niepożądanych wynosiła 17% w grupie PegIntron/REBETOL w porównaniu do 14% w grupie INTRON A/REBETOL.
wielu, ale nie we wszystkich przypadkach, działania niepożądane ustępowały po zmniejszeniu dawki lub przerwaniu leczenia. Niektórzy badani doświadczyli trwających lub nowych poważnych działań niepożądanych podczas 6-miesięcznego okresu obserwacji. W badaniu 2 wielu pacjentów nadal doświadczało działań niepożądanych kilka miesięcy po przerwaniu terapii. Pod koniec 6-miesięcznego okresu obserwacji częstość występowania utrzymujących się działań niepożądanych według grup ciała w grupie otrzymującej PegIntron 1,5/REBETOL 200 mg wynosiła 33% (psychiatryczne), 20% (mięśniowo-szkieletowe) i 10% (w obrębie układu hormonalnego i dla GI). U około 10 do 15% pacjentów utrata masy ciała, zmęczenie i ból głowy nie ustąpiły.
tych badaniach klinicznych odnotowano 31 zgonów pacjentów, które wystąpiły podczas leczenia lub podczas obserwacji. W badaniu 1 wystąpiło 1 samobójstwo u osoby otrzymującej PegIntron w monoterapii i 2 zgony wśród osób otrzymujących monoterapię INTRON A (1 morderstwo/samobójstwo i 1 nagły zgon). W badaniu 2 popełniono 1 samobójstwo u pacjenta otrzymującego terapię skojarzoną PegIntron/REBETOL 200 mg; oraz 1 zgon pacjenta w grupie INTRON A/REBETOL 200mg (wypadek samochodowy). W badaniu 3 było 14 zgonów, z których 2 były prawdopodobnymi samobójstwami, a 1 był niewyjaśnionym zgonem osoby z odpowiednią historią medyczną depresji. W badaniu 4 odnotowano 12 zgonów, z których 6 wystąpiło u pacjentów otrzymujących terapię skojarzoną PegIntron/REBETOL, 5 w ramieniu PegIntron 1,5 µg/REBETOL (N=1019) i 1 w ramieniu PegIntron 1 µg/REBETOL (N= 1016), a 6 z nich wystąpiło u osobników otrzymujących Pegasys/Copegus (N=1035); wystąpiły 3 samobójstwa, które miały miejsce w okresie obserwacji po leczeniu u pacjentów, którzy otrzymywali terapię skojarzoną PegIntron (1,5 µg/kg)/REBETOL 200 mg.
badaniach 1 i 2 od 10 do 14% pacjentów otrzymujących PegIntron w monoterapii lub w skojarzeniu z preparatem REBETOL 200 mg przerwało terapię, w porównaniu z 6% leczonych samym INTRON A i 13% leczonych INTRON A w skojarzeniu z REBETOLEM. Podobnie w Badaniu 3, 15% pacjentów otrzymujących PegIntron w skojarzeniu z zależnym od masy ciała produktem REBETOL i 14% pacjentów otrzymujących PegIntron i stałą dawkę REBETOL 200 mg przerwało terapię z powodu działania niepożądanego. Najczęstsze powody przerwania terapii były związane ze znanymi skutkami działań niepożądanych interferonu o charakterze psychiatrycznym, ogólnoustrojowym (np. zmęczenie, ból głowy) lub żołądkowo-jelitowymi. W badaniu 4 13% pacjentów w ramieniu PegIntron 1,5 µg/REBETOL 200 mg, 10% w ramieniu PegIntron 1 µg/REBETOL 200 mg i 13% w ramieniu Pegasys 180 µg/Copegus przerwało leczenie z powodu działań niepożądanych.
badaniu 2 zmniejszenie dawki z powodu działań niepożądanych wystąpiło u 42% pacjentów otrzymujących PegIntron (1,5 µg/kg)/REBETOL 200 mg i u 34% osób otrzymujących INTRON A/REBETOL. Większość pacjentów (57%) o masie ciała 60 kg lub mniejszej otrzymujących PegIntron (1,5 µg/kg)/REBETOL wymagała zmniejszenia dawki. Zmniejszenie stężenia interferonu było zależne od dawki (PegIntron 1,5 µg/kg większy niż PegIntron 0,5 µg/kg lub INTRON A), odpowiednio 40%, 27%, 28%. Redukcja dawki preparatu REBETOL 200 mg była podobna we wszystkich trzech grupach, od 33 do 35%. Najczęstszymi przyczynami modyfikacji dawki była neutropenia (18%) lub niedokrwistość (9%) (patrz Wartości laboratoryjne ). Inne częste przyczyny to depresja, zmęczenie, nudności i małopłytkowość. W badaniu 3 modyfikacje dawki z powodu działań niepożądanych występowały częściej przy dawkowaniu na podstawie masy ciała (WBD) w porównaniu z dawkowaniem stałym (odpowiednio 29% i 23%). W badaniu 4 u 16% pacjentów zmniejszono dawkę preparatu PegIntron do 1 μg/kg w skojarzeniu z preparatem REBETOL 200 mg, a dodatkowe 4% wymagało drugiego zmniejszenia dawki preparatu PegIntron do 0,5 μg/kg z powodu działań niepożądanych w porównaniu z 15% pacjentów w ramieniu Pegasys/Copegus, którzy wymagali zmniejszenia dawki do 135 μg/tydzień w przypadku Pegasys, a dodatkowe 7% w ramieniu Pegasys/Copegus wymagało drugiej redukcji dawki do 90 μg/tydzień w przypadku Pegasys.
badaniach skojarzonych PegIntron/REBETOL 200 mg najczęstszymi działaniami niepożądanymi były zaburzenia psychiczne, które wystąpiły u 77% uczestników badania 2 i 68% do 69% uczestników badania 3. Te psychiatryczne działania niepożądane obejmowały najczęściej depresję, drażliwość i bezsenność, każda zgłaszana przez około 30% do 40% pacjentów we wszystkich leczonych grupach. Zachowania samobójcze (wyobrażenia, próby i samobójstwa) wystąpiły u 2% wszystkich pacjentów w trakcie leczenia lub podczas obserwacji po zaprzestaniu leczenia [patrz OSTRZEŻENIA I ŚRODKI ]. W badaniu 4 psychiatryczne reakcje niepożądane wystąpiły u 58% pacjentów w ramieniu PegIntron 1,5 µg/REBETOL 200 mg, 55% pacjentów w ramieniu PegIntron 1 µg/REBETOL 200 mg i 57% pacjentów w ramieniu Pegasys 180 µg/Copegus .
PegIntron wywoływał zmęczenie lub ból głowy u około dwóch trzecich pacjentów, z gorączką lub dreszczami u około połowy pacjentów. Nasilenie niektórych z tych objawów ogólnoustrojowych (np. gorączka i ból głowy) zmniejszało się w miarę kontynuowania leczenia. W badaniach 1 i 2 zapalenie i reakcja w miejscu podania (np. siniak, swędzenie i podrażnienie) występowały z około dwukrotnie większą częstością w przypadku terapii PegIntron (do 75% pacjentów) w porównaniu z INTRON A. Jednak ból w miejscu wstrzyknięcia był rzadko (2 do 3%) we wszystkich grupach. W badaniu 3 ogólna częstość występowania reakcji w miejscu wstrzyknięcia lub stanu zapalnego wynosiła od 23% do 24%.
Pacjenci otrzymujący REBETOL/PegIntron w ramach ponownego leczenia po niepowodzeniu wcześniejszego schematu leczenia skojarzonego interferonem zgłaszali działania niepożądane podobne do tych, które były wcześniej związane z tym schematem podczas badań klinicznych u osób nieleczonych wcześniej.
Pacjenci pediatryczni
Ogólnie profil działań niepożądanych w populacji pediatrycznej był podobny do obserwowanego u dorosłych. W badaniu pediatrycznym najczęstszymi działaniami niepożądanymi u wszystkich pacjentów były: gorączka (80%), ból głowy (62%), neutropenia (33%), zmęczenie (30%), jadłowstręt (29%), rumień w miejscu wstrzyknięcia (29). %) i wymioty (27%). Większość działań niepożądanych zgłoszonych w badaniu miała nasilenie łagodne lub umiarkowane. Ciężkie działania niepożądane zgłoszono u 7% (8/107) wszystkich pacjentów i obejmowały ból w miejscu wstrzyknięcia (1%), ból kończyn (1%), ból głowy (1%), neutropenię (1%) i gorączkę (4 %). Ważnymi działaniami niepożądanymi, które wystąpiły w tej populacji pacjentów były nerwowość (7%; 7/107), agresja (3%; 3/107), gniew (2%; 2/107) i depresja (1%; 1/107) . Pięciu pacjentów otrzymało leczenie lewotyroksyną, trzech z kliniczną niedoczynnością tarczycy, a dwóch z bezobjawowym podwyższeniem TSH. Przyrost masy ciała i wzrostu dzieci leczonych preparatem PegIntron plus REBETOL pozostawał w tyle za przewidywanym na podstawie normatywnych danych populacyjnych przez cały okres leczenia. Poważnie zahamowaną prędkość wzrostu (mniej niż 3. percentyl) zaobserwowano u 70% badanych podczas leczenia.
Modyfikacje dawki produktu PegIntron i (lub) rybawiryny były wymagane u 25% pacjentów z powodu działań niepożądanych związanych z leczeniem, najczęściej związanych z niedokrwistością, neutropenią i utratą masy ciała. Dwóch pacjentów (2%; 2/107) przerwało leczenie w wyniku działania niepożądanego.
Działania niepożądane, które wystąpiły z częstością większą lub równą 10% u pacjentów pediatrycznych, przedstawiono w Tabeli 7.
Dziewięćdziesiąt cztery ze 107 pacjentów włączono do 5-letniego długoterminowego badania kontrolnego. Długoterminowy wpływ na wzrost był mniejszy u osób leczonych przez 24 tygodnie niż u osób leczonych przez 48 tygodni. Dwadzieścia cztery procent pacjentów (11/46) leczonych przez 24 tygodnie i 40% pacjentów (19/48) leczonych przez 48 tygodni miało >15 centylowy spadek wzrostu w zależności od wieku od okresu przed leczeniem do końca 5 roku obserwacja długoterminowa w porównaniu z percentylami wyjściowymi przed leczeniem. U jedenastu procent pacjentów (5/46) leczonych przez 24 tygodnie i u 13% pacjentów (6/48) leczonych przez 48 tygodni zaobserwowano spadek od wartości wyjściowych przed leczeniem o > 30 percentyli wzrostu w zależności od wieku do końca. z 5-letniego długoterminowego okresu obserwacji. Obserwowane we wszystkich grupach wiekowych, największe ryzyko zmniejszenia wzrostu pod koniec długoterminowej obserwacji wydaje się korelować z rozpoczęciem leczenia skojarzonego w latach spodziewanego szczytowego wzrostu. [Widzieć OSTRZEŻENIA I ŚRODKI ]
Wartości laboratoryjne
Osoby dorosłe i pediatryczne
Profil działań niepożądanych w badaniu 3, w którym porównano skojarzenie preparatu PegIntron/REBETOL 200 mg zależnym od masy ciała ze schematem leczenia skojarzonego PegIntron/REBETOL 200 mg o stałej dawce, wykazał zwiększoną częstość występowania niedokrwistości przy dawkowaniu zależnym od masy ciała (29% w porównaniu z 19% przy dawkowaniu zależnym od masy ciała). vs. schematy z płaską dawką). Jednak większość przypadków niedokrwistości była łagodna i reagowała na zmniejszenie dawki.
Poniżej opisano zmiany wybranych wartości laboratoryjnych podczas leczenia skojarzonego z preparatem REBETOL 200 mg. Zmniejszenie stężenia hemoglobiny, leukocytów, neutrofili i płytek krwi może wymagać zmniejszenia dawki lub trwałego przerwania leczenia [Widzieć DAWKOWANIE I SPOSÓB PODAWANIA ]. Zmiany wybranych wartości laboratoryjnych podczas leczenia przedstawiono w Tabeli 8. Większość zmian wartości laboratoryjnych w badaniu pediatrycznym PegIntron/REBETOL 200 mg była łagodna lub umiarkowana.
Hemoglobina
Stężenie hemoglobiny spadło do mniej niż 11 g/dl u około 30% uczestników badania 2. W badaniu 3 47% uczestników otrzymujących WBD REBETOL 200 mg i 33% stałą dawkę REBETOL 200 mg miało zmniejszenie stężenia hemoglobiny poniżej 11 g /dl. Zmniejszenie stężenia hemoglobiny do mniej niż 9 g/dl występowało częściej u pacjentów otrzymujących WBD w porównaniu z dawkowaniem stałym (odpowiednio 4% i 2%). W badaniu 2 modyfikacja dawki była wymagana u 9% i 13% pacjentów w grupach otrzymujących PegIntron/REBETOL 200 mg i INTRON A/REBETOL 200 mg. W badaniu 4 u pacjentów otrzymujących PegIntron (1,5 μg/kg)/REBETOL stwierdzono zmniejszenie stężenia hemoglobiny od 8,5 do mniej niż 10 g/dl (28%) i do mniej niż 8,5 g/dl (3%), podczas gdy u pacjentów otrzymujących Pegasys 180 µg/Copegus te spadki wystąpiły odpowiednio u 26% i 4% badanych. Poziomy hemoglobiny ustabilizowały się średnio po 4-6 tygodniach leczenia. Typowym zaobserwowanym wzorcem był spadek poziomu hemoglobiny w 4. tygodniu leczenia, po którym nastąpiła stabilizacja i plateau, które utrzymywały się do końca leczenia. W badaniu PegIntron monoterapii zmniejszenie stężenia hemoglobiny było na ogół łagodne i modyfikacje dawki były rzadko konieczne [patrz DAWKOWANIE I SPOSÓB PODAWANIA ].
Neutrofile
U większości dorosłych pacjentów leczonych terapią skojarzoną REBETOL 200 mg w Badaniu 2 (85%) i INTRON A/REBETOL (60%) zaobserwowano zmniejszenie liczby neutrofili. Ciężka, potencjalnie zagrażająca życiu neutropenia (mniej niż 0,5 x 109/l) wystąpiła u 2% pacjentów leczonych preparatem INTRON A/REBETOL 200 mg i u około 4% pacjentów leczonych preparatem PegIntron/REBETOL 200 mg w badaniu 2. Osiemnaście procent pacjentów otrzymujących PegIntron/REBETOL w Badaniu 2 wymagał modyfikacji dawkowania interferonu. Niewiele osób (mniej niż 1%) wymagało trwałego przerwania leczenia. Liczba neutrofili zazwyczaj powracała do poziomu sprzed leczenia 4 tygodnie po zaprzestaniu leczenia [patrz DAWKOWANIE I SPOSÓB PODAWANIA ].
Płytki krwi
Liczba płytek krwi zmniejszyła się do mniej niż 100 000/mm3 u około 20% pacjentów leczonych samym PegIntronem lub preparatem REBETOL 200 mg oraz u 6% pacjentów dorosłych leczonych preparatem INTRON A/REBETOL. Ciężkie zmniejszenie liczby płytek krwi (poniżej 50 000/mm³) występuje u mniej niż 4% dorosłych osób. Pacjenci mogą wymagać przerwania leczenia lub modyfikacji dawki w wyniku zmniejszenia liczby płytek krwi [patrz DAWKOWANIE I SPOSÓB PODAWANIA ]. W Badaniu 2 1% lub 3% uczestników wymagało modyfikacji dawki odpowiednio INTRON A lub PegIntron. Liczba płytek krwi na ogół powracała do poziomu sprzed leczenia 4 tygodnie po zakończeniu terapii.
Funkcja tarczycy
Rozwój nieprawidłowości TSH, z objawami klinicznymi lub bez, jest związany z terapiami interferonowymi. W badaniu 2 klinicznie widoczne zaburzenia tarczycy występowały u pacjentów leczonych preparatem INTRON A lub PegIntron (z lub bez REBETOL) z podobną częstością (5% w przypadku niedoczynności tarczycy i 3% w przypadku nadczynności tarczycy). U badanych pojawiły się nowe nieprawidłowości TSH o początku podczas leczenia i w okresie obserwacji. Pod koniec okresu obserwacji 7% badanych nadal miało nieprawidłowe wartości TSH.
Bilirubina i kwas moczowy
W badaniu 2 10 do 14% pacjentów rozwinęła hiperbilirubinemię, a od 33 do 38% hiperurykemię związaną z hemolizą. U sześciu pacjentów wystąpiła dna moczanowa o nasileniu łagodnym do umiarkowanego.
Doświadczenie w badaniach klinicznych – Terapia skojarzona REBETOL/INTRON A
Osoby dorosłe
badaniach klinicznych odpowiednio 19% i 6% pacjentów wcześniej nieleczonych i pacjentów z nawrotem przerwało leczenie z powodu działań niepożądanych w ramionach leczenia skojarzonego w porównaniu z 13% i 3% w ramionach z interferonem. Wybrane działania niepożądane związane z leczeniem, które wystąpiły w badaniach w USA z częstością większą lub równą 5%, przedstawiono według grup leczenia (patrz Tabela 9). Na ogół wybrane działania niepożądane związane z leczeniem zgłaszano z mniejszą częstością w badaniach międzynarodowych w porównaniu z badaniami w USA, z wyjątkiem osłabienia, objawów grypopodobnych, nerwowości i świądu.
Pacjenci pediatryczni
badaniach klinicznych z udziałem 118 dzieci w wieku od 3 do 16 lat 6% przerwało leczenie z powodu działań niepożądanych. Modyfikacje dawki były wymagane u 30% badanych, najczęściej z powodu niedokrwistości i neutropenii. Ogólnie profil działań niepożądanych w populacji pediatrycznej był podobny do obserwowanego u dorosłych. Zaburzenia w miejscu wstrzyknięcia, gorączka, anoreksja, wymioty i chwiejność emocjonalna występowały częściej u pacjentów pediatrycznych w porównaniu z osobami dorosłymi. Odwrotnie, dzieci i młodzież odczuwały mniejsze zmęczenie, niestrawność, bóle stawów, bezsenność, drażliwość, zaburzenia koncentracji, duszność i świąd w porównaniu z osobami dorosłymi. Wybrane działania niepożądane związane z leczeniem, które wystąpiły z częstością większą lub równą 5% wśród wszystkich dzieci, które otrzymały zalecaną dawkę terapii skojarzonej REBETOL/INTRON A, przedstawiono w Tabeli 9.
trakcie 48-tygodniowej terapii nastąpił spadek szybkości liniowego wzrostu (średni spadek przypisania centylowego o 7%) oraz spadek szybkości przyrostu masy ciała (średni spadek przypisania centylowego o 9%). Ogólne odwrócenie tych trendów odnotowano w ciągu 24-tygodniowego okresu po leczeniu. Jednak długoterminowe dane z ograniczonej liczby pacjentów sugerują, że terapia skojarzona może wywoływać zahamowanie wzrostu, co skutkuje obniżonym ostatecznym wzrostem u niektórych pacjentów [patrz OSTRZEŻENIA I ŚRODKI ].
Wartości laboratoryjne
Poniżej opisano zmiany wybranych wartości hematologicznych (hemoglobiny, krwinek białych, neutrofili i płytek krwi) podczas leczenia (patrz Tabela 10).
Hemoglobina. Spadek stężenia hemoglobiny wśród pacjentów otrzymujących terapię REBETOL rozpoczął się w 1. tygodniu, ze stabilizacją do 4. tygodnia. test. U pacjentów z nawrotem średni maksymalny spadek w stosunku do wartości wyjściowej wynosił 2,8 g/dl w badaniu amerykańskim i 2,6 g/dl w badaniu międzynarodowym. U większości pacjentów wartości hemoglobiny powróciły do poziomów sprzed leczenia w ciągu 4 do 8 tygodni od zakończenia terapii.
Bilirubina i kwas moczowy. W badaniach klinicznych odnotowano wzrost zarówno bilirubiny, jak i kwasu moczowego, związany z hemolizą. Większość z nich była umiarkowanymi zmianami biochemicznymi, które ustąpiły w ciągu 4 tygodni po przerwaniu leczenia. Ta obserwacja występowała najczęściej u osób z wcześniejszym rozpoznaniem zespołu Gilberta. Nie było to związane z zaburzeniami czynności wątroby lub chorobowością kliniczną.
Doświadczenia postmarketingowe
Następujące działania niepożądane zostały zidentyfikowane i zgłoszone podczas stosowania produktu REBETOL po dopuszczeniu do obrotu w połączeniu z INTRON A lub PegIntron. Ponieważ reakcje te są zgłaszane dobrowolnie w populacji o niepewnej wielkości, nie zawsze jest możliwe wiarygodne oszacowanie ich częstości lub ustalenie związku przyczynowego z ekspozycją na lek.
Zaburzenia krwi i układu chłonnego
Czysta aplazja czerwonokrwinkowa, niedokrwistość aplastyczna
Zaburzenia ucha i labiryntu
zaburzenia słuchu, zawroty głowy
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia
Nadciśnienie płucne
Zaburzenia oka
Poważne odwarstwienie siatkówki
Zaburzenia endokrynologiczne
Cukrzyca
INTERAKCJE Z LEKAMI
Didanozyna
Ekspozycja na didanozynę lub jej aktywny metabolit (5'-trifosforan dideoksyadenozyny) jest zwiększona, gdy didanozyna jest podawana jednocześnie z rybawiryną, co może powodować lub nasilać toksyczność kliniczną; dlatego jednoczesne podawanie kapsułek lub roztworu doustnego REBETOL 200 mg i didanozyny jest przeciwwskazane. W badaniach klinicznych zgłaszano przypadki śmiertelnej niewydolności wątroby, neuropatii obwodowej, zapalenia trzustki i objawowej hiperlaktatemii/kwasicy mleczanowej.
Analogi nukleozydów
Dekompensacja czynności wątroby (niektóre śmiertelne) wystąpiła u pacjentów z marskością wątroby jednocześnie zakażonych HIV/HCV, otrzymujących skojarzoną terapię przeciwretrowirusową HIV z interferonem alfa i rybawiryną. Dodanie leczenia interferonami alfa w monoterapii lub w skojarzeniu z rybawiryną może zwiększyć ryzyko w tej populacji pacjentów. Pacjenci otrzymujący interferon z rybawiryną i nukleozydowymi inhibitorami odwrotnej transkryptazy (NRTI) powinni być ściśle monitorowani pod kątem toksyczności związanej z leczeniem, zwłaszcza dekompensacji czynności wątroby i niedokrwistości. Przerwanie stosowania NRTI należy rozważyć z medycznego punktu widzenia (patrz oznakowanie indywidualnego produktu NRTI ). Zmniejszenie dawki lub odstawienie interferonu, rybawiryny lub obu tych leków należy również rozważyć w przypadku zaobserwowania pogorszenia toksyczności klinicznej, w tym dekompensacji czynności wątroby (np. wartość Child-Pugh większa niż 6).
Rybawiryna może antagonizować przeciwwirusowe działanie stawudyny i zydowudyny w hodowli komórkowej na HIV. Wykazano, że rybawiryna w hodowli komórkowej hamuje fosforylację lamiwudyny, stawudyny i zydowudyny, co może prowadzić do zmniejszenia aktywności przeciwretrowirusowej. Jednak w badaniu z innym pegylowanym interferonem w skojarzeniu z rybawiryną nie zaobserwowano interakcji farmakokinetycznych (np. stężenia w osoczu lub wewnątrzkomórkowych stężeniach metabolitów trifosforylowanego aktywnego metabolitu) ani farmakodynamicznych (np. utrata supresji wirusologicznej HIV/HCV), gdy rybawiryna i lamiwudyna (n =18), stawudynę (n=10) lub zydowudynę (n=6) podawano jednocześnie jako część schematu wielolekowego u pacjentów jednocześnie zakażonych HIV/HCV. Dlatego należy zachować ostrożność podczas jednoczesnego stosowania rybawiryny z którymkolwiek z tych leków.
Leki metabolizowane przez cytochrom P-450
Wyniki badań in vitro z użyciem preparatów mikrosomów wątroby ludzkiej i szczurzej wykazały niewielki lub brak metabolizmu rybawiryny za pośrednictwem enzymów cytochromu P-450, przy minimalnym potencjale interakcji lekowych opartych na enzymach P-450.
Nie zaobserwowano interakcji farmakokinetycznych pomiędzy kapsułkami INTRON A i REBETOL 200 mg w badaniu farmakokinetyki wielokrotnej dawki.
Azatiopryna
Istnieją doniesienia, że stosowanie rybawiryny w leczeniu przewlekłego wirusowego zapalenia wątroby typu C u pacjentów otrzymujących azatioprynę wywołuje ciężką pancytopenię i może zwiększać ryzyko mielotoksyczności związanej z azatiopryną. Dehydrogenaza monofosforanu inozyny (IMDH) jest wymagana dla jednego ze szlaków metabolicznych azatiopryny. Wiadomo, że rybawiryna hamuje IMDH, prowadząc w ten sposób do akumulacji metabolitu azatiopryny, 6-metylotioinozynomonofosforanu (6-MTITP), co jest związane z mielotoksycznością (neutropenia, trombocytopenia i niedokrwistość). Pacjenci otrzymujący azatioprynę z rybawiryną powinni mieć pełną morfologię krwi, w tym liczbę płytek krwi, monitorowaną co tydzień przez pierwszy miesiąc, dwa razy w miesiącu w drugim i trzecim miesiącu leczenia, a następnie co miesiąc lub częściej, jeśli konieczne są zmiany dawkowania lub inne leczenie [patrz OSTRZEŻENIA I ŚRODKI ].
OSTRZEŻENIA
Zawarte jako część ŚRODKI OSTROŻNOŚCI Sekcja.
ŚRODKI OSTROŻNOŚCI
Ciąża
Kapsułki REBETOL 200 mg i roztwór doustny mogą powodować wady wrodzone i śmierć nienarodzonego dziecka. Leczenie preparatem REBETOL nie powinno być rozpoczynane przed uzyskaniem negatywnego wyniku testu ciążowego bezpośrednio przed planowanym rozpoczęciem leczenia. Pacjentki powinny stosować co najmniej dwie formy antykoncepcji i wykonywać comiesięczne testy ciążowe w trakcie leczenia oraz w okresie 6 miesięcy po zakończeniu leczenia. Należy zachować szczególną ostrożność, aby uniknąć ciąży u pacjentek i partnerek pacjentów płci męskiej. Wykazał REBETOL 200mg znaczące działanie teratogenne i embriobójcze u wszystkich gatunków zwierząt, na których przeprowadzono odpowiednie badania. Efekty te wystąpiły przy dawkach, jak na poziomie jednej dwudziestej zalecanej dawki rybawiryny stosowanej u ludzi. Nie należy rozpoczynać terapii REBETOL 200 mg do czasu uzyskania negatywnego wyniku testu ciążowego został uzyskany bezpośrednio przed planowanym rozpoczęciem leczenia [patrz OSTRZEŻENIE W PUDEŁKU , PRZECIWWSKAZANIA , Używaj w określonych populacjach , oraz INFORMACJA O PACJENCIE ].
Niedokrwistość
Pierwotną toksycznością rybawiryny jest niedokrwistość hemolityczna, którą obserwowano u około 10% pacjentów leczonych preparatem REBETOL/INTRON A w badaniach klinicznych. Niedokrwistość związana z kapsułkami REBETOL występuje w ciągu 1 do 2 tygodni od rozpoczęcia leczenia. Ponieważ początkowy spadek stężenia hemoglobiny może być znaczny, zaleca się oznaczenie stężenia hemoglobiny lub hematokrytu przed rozpoczęciem leczenia oraz w 2. i 4. tygodniu leczenia lub częściej, jeśli jest to wskazane klinicznie. Pacjenci powinni być następnie obserwowani, jeśli jest to klinicznie uzasadnione [patrz DAWKOWANIE I SPOSÓB PODAWANIA ].
pacjentów z niedokrwistością wywołaną przez REBETOL zgłaszano przypadki zawałów mięśnia sercowego zakończone zgonem i niezakończone zgonem. Przed rozpoczęciem leczenia rybawiryną pacjentów należy ocenić pod kątem podstawowej choroby serca. Pacjenci z istniejącą wcześniej chorobą serca powinni mieć wykonane elektrokardiogramy przed leczeniem i powinni być odpowiednio monitorowani podczas leczenia. W przypadku jakiegokolwiek pogorszenia stanu sercowo-naczyniowego leczenie należy przerwać lub przerwać [patrz DAWKOWANIE I SPOSÓB PODAWANIA ]. Ponieważ choroba serca może ulec pogorszeniu przez niedokrwistość polekową, pacjenci z historią poważnej lub niestabilnej choroby serca nie powinni stosować REBETOL.
Zapalenie trzustki
Leczenie preparatami REBETOL i INTRON A lub PegIntron należy przerwać u pacjentów z objawami przedmiotowymi i podmiotowymi zapalenia trzustki, a przerwać u pacjentów z potwierdzonym zapaleniem trzustki.
Choroby płuc
Podczas leczenia produktem REBETOL z terapią skojarzoną interferonem alfa zgłaszano objawy płucne, w tym duszność, nacieki w płucach, zapalenie płuc, nadciśnienie płucne i zapalenie płuc; zdarzały się sporadyczne przypadki śmiertelnego zapalenia płuc. Ponadto zgłaszano sarkoidozę lub zaostrzenie sarkoidozy. Jeśli istnieją dowody na nacieki w płucach lub zaburzenia czynności płuc, pacjenta należy ściśle monitorować iw razie potrzeby przerwać leczenie skojarzone.
Zaburzenia okulistyczne
Rybawiryna jest stosowana w terapii skojarzonej z interferonami alfa. Pogorszenie lub utrata wzroku, retinopatia, w tym obrzęk plamki, tętnica lub żyła siatkówki, zakrzepica, krwotoki siatkówkowe i plamy waty, zapalenie nerwu wzrokowego, obrzęk tarczy nerwu wzrokowego i surowicze odwarstwienie siatkówki są wywoływane lub pogarszane przez leczenie interferonami alfa. Wszyscy pacjenci powinni otrzymać badanie wzroku przed rozpoczęciem leczenia. Pacjenci z istniejącymi wcześniej zaburzeniami okulistycznymi (np. retinopatią cukrzycową lub nadciśnieniową) powinni być poddawani okresowym badaniom okulistycznym podczas leczenia skojarzonego z interferonem alfa. Każdy pacjent, u którego wystąpią objawy oczne, powinien otrzymać szybkie i pełne badanie oczu. Leczenie skojarzone z interferonami alfa należy przerwać u pacjentów, u których wystąpią nowe zaburzenia okulistyczne lub ich nasilenie.
Testy laboratoryjne
PegIntron w połączeniu z rybawiryną może powodować poważne zmniejszenie liczby neutrofili i płytek krwi oraz zaburzenia hematologiczne, endokrynologiczne (np. TSH) i wątrobowe.
Pacjenci stosujący terapię skojarzoną PegIntron/REBETOL powinni być poddawani badaniom hematologicznym i biochemicznym krwi przed rozpoczęciem leczenia, a następnie okresowo. W badaniu klinicznym z udziałem dorosłych w okresie leczenia w tygodniach 2, 4, 8, 12 i następnie w odstępach 6-tygodniowych lub częściej w przypadku pojawienia się nieprawidłowości. U pacjentów pediatrycznych te same parametry laboratoryjne oceniano z dodatkową oceną stężenia hemoglobiny w 6. tygodniu leczenia. Poziomy TSH mierzono co 12 tygodni w okresie leczenia. W trakcie leczenia należy okresowo oznaczać HCV-RNA [patrz DAWKOWANIE I SPOSÓB PODAWANIA ].
Choroby stomatologiczne i przyzębia
pacjentów otrzymujących rybawirynę i interferon lub peginterferon w leczeniu skojarzonym zgłaszano zaburzenia zębów i przyzębia. Ponadto suchość w jamie ustnej może mieć szkodliwy wpływ na zęby i błony śluzowe jamy ustnej podczas długotrwałego leczenia kombinacją REBETOL i pegylowanego lub niepegylowanego interferonu alfa-2b. Pacjenci powinni dokładnie myć zęby dwa razy dziennie i poddawać się regularnym badaniom stomatologicznym. W przypadku wystąpienia wymiotów należy im zalecić dokładne wypłukanie ust.
Jednoczesne podawanie azatiopryny
piśmiennictwie opisywano pancytopenię (znaczne zmniejszenie liczby czerwonych krwinek, neutrofili i płytek krwi) oraz zahamowanie czynności szpiku kostnego w ciągu 3 do 7 tygodni po jednoczesnym podaniu pegylowanego interferonu/rybawiryny i azatiopryny. W tej ograniczonej liczbie pacjentów (n=8) mielotoksyczność była odwracalna w ciągu 4 do 6 tygodni po odstawieniu zarówno leczenia przeciwwirusowego HCV, jak i jednoczesnego leczenia azatiopryną i nie nawracała po ponownym włączeniu obu leków. PegIntron, REBETOL i azatioprynę należy odstawić w przypadku pancytopenii i nie należy ponownie wprowadzać pegylowanego interferonu z rybawiryną jednocześnie z azatiopryną [patrz INTERAKCJE Z LEKAMI ].
Wpływ na wzrost — zastosowanie pediatryczne
Dane dotyczące wpływu PegIntron i REBETOL na wzrost pochodzą z otwartego badania z udziałem osób w wieku od 3 do 17 lat, w którym zmiany masy ciała i wzrostu porównywano z normatywnymi danymi populacyjnymi w USA. Ogólnie rzecz biorąc, przyrost masy ciała i wzrostu dzieci leczonych preparatami PegIntron i REBETOL 200 mg pozostaje w tyle za przewidywanym na podstawie normatywnych danych populacyjnych przez cały okres leczenia. Poważnie zahamowaną prędkość wzrostu (mniej niż 3. percentyl) zaobserwowano u 70% badanych podczas leczenia. Po leczeniu u większości pacjentów wystąpił wzrost z odbicia i przyrost masy ciała. Dane z długoterminowej obserwacji pacjentów pediatrycznych wskazują jednak, że PegIntron w terapii skojarzonej z produktem REBETOL może indukować zahamowanie wzrostu, co skutkuje zmniejszeniem wzrostu dorosłego u niektórych pacjentów [patrz DZIAŁANIA NIEPOŻĄDANE ].
Podobnie, wpływ na wzrost zaobserwowano u pacjentów po leczeniu skojarzonym REBETOL 200 mg i INTRON A przez jeden rok. W długoterminowym badaniu obserwacyjnym na ograniczonej liczbie tych pacjentów, terapia skojarzona doprowadziła u niektórych pacjentów do zmniejszenia końcowego wzrostu osoby dorosłej [patrz DZIAŁANIA NIEPOŻĄDANE ].
Zabezpieczenia użytkowania
Na podstawie wyników badań klinicznych monoterapia rybawiryną nie jest skuteczna w leczeniu przewlekłego zakażenia wirusem zapalenia wątroby typu C; dlatego nie wolno stosować samodzielnie kapsułek lub roztworu doustnego REBETOL 200 mg. Bezpieczeństwo i skuteczność kapsułek i roztworu doustnego REBETOL ustalono jedynie w przypadku stosowania razem z INTRON A lub PegIntron (nie innymi interferonami) w terapii skojarzonej.
Bezpieczeństwo i skuteczność terapii REBETOL/INTRON A i PegIntron w leczeniu zakażenia HIV, adenowirusem, RSV, paragrypą lub grypą nie zostały ustalone. W tych wskazaniach nie należy stosować kapsułek REBETOL 200 mg. Rybawiryna do inhalacji posiada oddzielne oznakowanie, z którym należy zapoznać się, jeśli rozważana jest terapia wziewna rybawiryną.
Istnieją znaczące działania niepożądane spowodowane terapią REBETOL/INTRON A lub PegIntron, w tym ciężka depresja i myśli samobójcze, niedokrwistość hemolityczna, zahamowanie czynności szpiku kostnego, zaburzenia autoimmunologiczne i zakaźne, dysfunkcja płuc, zapalenie trzustki i cukrzyca. Myśli lub próby samobójcze występowały częściej u pacjentów pediatrycznych, głównie młodzieży, w porównaniu z pacjentami dorosłymi (2,4% w porównaniu z 1%) w trakcie leczenia i obserwacji po zakończeniu terapii. Przed rozpoczęciem leczenia skojarzonego należy w całości przejrzeć oznakowanie produktów INTRON A i PegIntron w celu uzyskania dodatkowych informacji dotyczących bezpieczeństwa.
Informacje dotyczące poradnictwa dla pacjentów
Poinformuj pacjenta, aby przeczytał zatwierdzone przez FDA oznakowanie pacjenta ( Przewodnik po lekach ).
Niedokrwistość
Najczęstszym działaniem niepożądanym występującym podczas stosowania kapsułek REBETOL jest niedokrwistość, która może być ciężka [patrz OSTRZEŻENIA I ŚRODKI oraz DZIAŁANIA NIEPOŻĄDANE ]. Pacjentów należy pouczyć, że przed rozpoczęciem leczenia, a następnie okresowo, wymagane są badania laboratoryjne [patrz DAWKOWANIE I SPOSÓB PODAWANIA ]. Wskazane jest, aby pacjenci byli dobrze nawodnieni, zwłaszcza na początkowych etapach leczenia.
Ciąża
Należy poinformować pacjentów, że kapsułki REBETOL 200 mg i roztwór doustny mogą powodować wady wrodzone i śmierć nienarodzonego dziecka. REBETOL 200mg nie może być stosowany przez kobiety w ciąży lub przez mężczyzn, których partnerki są w ciąży. Należy zachować szczególną ostrożność, aby uniknąć ciąży u pacjentek i partnerek pacjentów płci męskiej przyjmujących REBETOL. Nie należy rozpoczynać stosowania preparatu REBETOL do czasu uzyskania negatywnego wyniku testu ciążowego bezpośrednio przed rozpoczęciem leczenia. Pacjentki muszą wykonywać test ciążowy co miesiąc podczas terapii i przez 6 miesięcy po terapii.
Kobiety w wieku rozrodczym muszą być poinformowane o stosowaniu skutecznej antykoncepcji (dwie niezawodne formy) przed rozpoczęciem leczenia. Pacjentki (mężczyźni i kobiety) muszą zostać poinformowane o ryzyku teratogennym/zarodkowym i muszą być poinstruowane o stosowaniu skutecznej antykoncepcji podczas stosowania preparatu REBETOL 200 mg i przez 6 miesięcy po zakończeniu leczenia. Pacjentom (mężczyznom i kobietom) należy zalecić natychmiastowe powiadomienie lekarza w przypadku zajścia w ciążę [patrz PRZECIWWSKAZANIA , OSTRZEŻENIA I ŚRODKI , oraz Stosowanie w określonych populacjach ].
W przypadku zajścia w ciążę podczas leczenia lub w ciągu 6 miesięcy po zakończeniu leczenia, pacjentkę należy poinformować o ryzyku działania teratogennego na płód po zastosowaniu preparatu REBETOL 200 mg. Pacjenci lub partnerzy pacjentów powinni niezwłocznie zgłaszać lekarzowi każdą ciążę, która wystąpiła podczas leczenia lub w ciągu 6 miesięcy po zakończeniu leczenia. Osoby przepisujące receptę powinny zgłaszać takie przypadki dzwoniąc pod numer 1-800-593-2214.
Ryzyko kontra korzyści
Pacjenci otrzymujący kapsułki REBETOL powinni być poinformowani o korzyściach i zagrożeniach związanych z leczeniem, ukierunkowanym na jego właściwe stosowanie i skierowanym do pacjenta Przewodnik po lekach . Należy poinformować pacjentów, że wpływ leczenia zakażenia wirusem zapalenia wątroby typu C na przenoszenie nie jest znany i że należy podjąć odpowiednie środki ostrożności zapobiegające przenoszeniu wirusa zapalenia wątroby typu C.
Pacjentów należy poinformować o tym, co zrobić w przypadku pominięcia dawki leku REBETOL; pominiętą dawkę należy przyjąć jak najszybciej tego samego dnia. Pacjenci nie powinni podwajać następnej dawki. Pacjentom należy doradzić, aby w razie pytań skontaktowali się ze swoim lekarzem.
Toksykologia niekliniczna
rakotwórczość, mutageneza, upośledzenie płodności
Karcynogeneza
Rybawiryna podawana przez 6 miesięcy w modelu myszy transgenicznych z niedoborem p53 w dawkach do 300 mg/kg nie powodowała wzrostu liczby guzów (szacunkowy ludzki ekwiwalent 25 mg/kg w oparciu o korektę powierzchni ciała dla osoby dorosłej o masie 60 kg). około 1,9-krotność maksymalnej zalecanej dawki dobowej dla ludzi). Rybawiryna nie działała rakotwórczo, gdy była podawana szczurom przez 2 lata w dawkach do 40 mg/kg (szacunkowy ekwiwalent u ludzi 5,71 mg/kg na podstawie dostosowania powierzchni ciała dla osoby dorosłej o masie 60 kg).
Mutageneza
Rybawiryna wykazała zwiększoną częstość występowania mutacji i transformacji komórek w wielu testach genotoksyczności. Rybawiryna była aktywna w teście transformacji komórek Balb/3T3 in vitro. Aktywność mutagenną zaobserwowano w teście chłoniaka myszy i przy dawkach od 20 do 200 mg/kg (szacowany ekwiwalent u ludzi wynoszący 1,67 do 16,7 mg/kg, w oparciu o dostosowanie powierzchni ciała dla osoby dorosłej o masie 60 kg; 0,1 do 1 razy większej od maksymalnej zalecanej 24-godzinnej dawki rybawiryny u ludzi) w teście mikrojądrowym u myszy. Dominujący test śmiertelności u szczurów był negatywny, co wskazuje, że jeśli mutacje wystąpiły u szczurów, nie były one przenoszone przez gamety męskie.
Upośledzenie płodności
Rybawiryna wykazywała znaczące działanie embriobójcze i teratogenne w dawkach znacznie niższych od zalecanej dawki dla ludzi u wszystkich gatunków zwierząt, na których przeprowadzono odpowiednie badania. Stwierdzono wady rozwojowe czaszki, podniebienia, oka, szczęki, kończyn, szkieletu i przewodu pokarmowego. Częstość występowania i nasilenie efektów teratogennych wzrastały wraz ze wzrostem dawki leku. Przeżywalność płodów i potomstwa została zmniejszona. W konwencjonalnych badaniach embriotoksyczności/teratogenności u szczurów i królików, obserwowane poziomy dawek niepowodujących zmian były znacznie niższe od proponowanych do stosowania klinicznego (0,3 mg/kg/dobę zarówno dla szczurów, jak i królików; około 0,06-krotność zalecanej dawki dobowej u ludzi rybawiryna). Nie zaobserwowano toksyczności matczynej ani wpływu na potomstwo w badaniu toksyczności około- i poporodowej u szczurów, którym podawano doustnie do 1 mg/kg mc./dobę (szacunkowa dawka ekwiwalentna u ludzi wynosząca 0,17 mg/kg w oparciu o korektę powierzchni ciała u dorosłego osobnika o masie 60 kg). około 0,01-krotność maksymalnej zalecanej 24-godzinnej dawki rybawiryny stosowanej u ludzi) [patrz PRZECIWWSKAZANIA , oraz OSTRZEŻENIA I ŚRODKI ].
Kobiety płodne i partnerzy kobiet płodnych nie powinny otrzymywać leku REBETOL, chyba że pacjent i jego partner stosują skuteczną antykoncepcję (dwie niezawodne formy). Biorąc pod uwagę okres półtrwania wielokrotnej dawki (t1/2) rybawiryny wynoszący 12 dni, skuteczna antykoncepcja musi być stosowana przez 6 miesięcy po leczeniu (np. 15 okresów półtrwania dla klirensu rybawiryny).
REBETOL należy stosować ostrożnie u płodnych mężczyzn. W badaniach na myszach mających na celu ocenę przebiegu czasowego i odwracalności zwyrodnienia jąder wywołanego rybawiryną w dawkach od 15 do 150 mg/kg/dobę (szacunkowy ekwiwalent u ludzi 1,25 do 12,5 mg/kg/dobę, na podstawie korekty powierzchni ciała dla Dorosły osobnik o masie ciała 60 kg; 0,1-0,8-krotność maksymalnej dobowej dawki rybawiryny u ludzi) podawanych przez 3 lub 6 miesięcy, wystąpiły nieprawidłowości w nasieniu. Po zaprzestaniu leczenia, zasadniczo całkowite wyleczenie wywołanej rybawiryną toksyczności jąder było widoczne w ciągu 1 lub 2 cykli spermatogenezy.
Używaj w określonych populacjach
Ciąża
Kategoria ciąży X
[Widzieć PRZECIWWSKAZANIA , OSTRZEŻENIA I ŚRODKI , oraz Toksykologia niekliniczna ].
Leczenie i po leczeniu:
Potencjalne ryzyko dla płodu
Wiadomo, że rybawiryna gromadzi się w składnikach wewnątrzkomórkowych, skąd jest bardzo powoli usuwana. Nie wiadomo, czy rybawiryna zawarta w nasieniu będzie wywierać potencjalny wpływ teratogenny na zapłodnienie komórki jajowej. W badaniu na szczurach stwierdzono, że rybawiryna w dawkach do 200 mg/kg przez 5 dni nie wywołała dominującej śmiertelności (szacunkowa dawka u ludzi wynosi od 7,14 do 28,6 mg/kg, na podstawie skorygowanej powierzchni ciała dla 60 kg dorosły; do 1,7-krotności maksymalnej zalecanej dawki rybawiryny dla ludzi). Jednak ze względu na potencjalne działanie teratogenne rybawiryny u ludzi, mężczyznom należy zalecić podjęcie wszelkich środków ostrożności w celu uniknięcia ryzyka zajścia w ciążę u partnerek.
Kobiety w wieku rozrodczym nie powinny otrzymywać REBETOL, chyba że stosują skuteczną antykoncepcję (dwie niezawodne formy) w okresie leczenia. Ponadto, skuteczna antykoncepcja powinna być stosowana przez 6 miesięcy po leczeniu w oparciu o okres półtrwania wielokrotnej dawki (t1/2) rybawiryny wynoszący 12 dni.
Mężczyźni i ich partnerki muszą stosować skuteczną antykoncepcję (dwie niezawodne formy) podczas leczenia produktem REBETOL i przez 6 miesięcy po terapii (np. 15 okresów półtrwania dla klirensu rybawiryny z organizmu).
Utworzono rejestr ciążowy produktu Ribavirin Pregnancy Registry w celu monitorowania wyników ciąży matek i płodu u pacjentek i partnerek pacjentów płci męskiej narażonych na działanie rybawiryny w trakcie leczenia i przez 6 miesięcy po jego zakończeniu. Zachęcamy lekarzy i pacjentów do zgłaszania takich przypadków pod numerem telefonu 1-800-593-2214.
Matki karmiące
Nie wiadomo, czy produkt REBETOL przenika do mleka ludzkiego. Ze względu na możliwość wystąpienia poważnych działań niepożądanych leku u niemowląt karmionych piersią, należy podjąć decyzję, czy przerwać karmienie piersią, czy opóźnić lub przerwać stosowanie preparatu REBETOL.
Zastosowanie pediatryczne
Bezpieczeństwo i skuteczność preparatu REBETOL 200 mg w skojarzeniu z PegIntronem nie zostały ustalone u dzieci w wieku poniżej 3 lat. W przypadku leczenia preparatem REBETOL/INTRON A, podejmując decyzję o leczeniu pacjenta pediatrycznego, należy wziąć pod uwagę dowody progresji choroby, takie jak zapalenie i zwłóknienie wątroby, a także czynniki prognostyczne odpowiedzi, genotyp HCV i miano wirusa. Korzyści z leczenia należy porównać z zaobserwowanymi wynikami dotyczącymi bezpieczeństwa.
Dane z długoterminowej obserwacji pacjentów pediatrycznych wskazują, że REBETOL w skojarzeniu z PegIntronem lub INTRON A może indukować zahamowanie wzrostu, co skutkuje zmniejszeniem wzrostu u niektórych pacjentów [patrz OSTRZEŻENIA I ŚRODKI oraz DZIAŁANIA NIEPOŻĄDANE ].
Myśli lub próby samobójcze występowały częściej u pacjentów pediatrycznych, głównie młodzieży, w porównaniu z pacjentami dorosłymi (2,4% w porównaniu z 1%) w trakcie leczenia i obserwacji po zakończeniu terapii [Widzieć OSTRZEŻENIA I ŚRODKI ]. Podobnie jak u pacjentów dorosłych, u dzieci i młodzieży wystąpiły inne psychiatryczne reakcje niepożądane (np. depresja, labilność emocjonalna, senność), niedokrwistość i neutropenia [patrz OSTRZEŻENIA I ŚRODKI ].
Zastosowanie geriatryczne
Badania kliniczne terapii REBETOL/INTRON A lub PegIntron nie obejmowały wystarczającej liczby osób w wieku 65 lat i starszych, aby ustalić, czy reagują oni inaczej niż osoby młodsze.
Wiadomo, że REBETOL 200 mg jest w znacznym stopniu wydalany przez nerki, a ryzyko reakcji toksycznych na ten lek może być większe u pacjentów z zaburzeniami czynności nerek. Ponieważ pacjenci w podeszłym wieku często mają zmniejszoną czynność nerek, należy zachować ostrożność przy doborze dawki. Należy monitorować czynność nerek i odpowiednio dostosować dawkowanie. REBETOL 200 mg nie powinien być stosowany u pacjentów z klirensem kreatyniny mniejszym niż 50 ml/min [patrz PRZECIWWSKAZANIA ].
Ogólnie rzecz biorąc, kapsułki REBETOL 200 mg należy podawać pacjentom w podeszłym wieku z ostrożnością, zaczynając od dolnego zakresu dawkowania, co odzwierciedla większą częstość występowania pogorszenia czynności wątroby i serca oraz współistniejących chorób lub innych leków. W badaniach klinicznych osoby w podeszłym wieku częściej miały niedokrwistość (67%) niż młodsi pacjenci (28%) [patrz OSTRZEŻENIA I ŚRODKI ].
Biorcy przeszczepów narządów
Bezpieczeństwo i skuteczność INTRON A i PegIntron w monoterapii lub w skojarzeniu z REBETOL w leczeniu zapalenia wątroby typu C u biorców wątroby lub innych narządów nie zostały ustalone. W małym (n = 16) jednoośrodkowym, niekontrolowanym doświadczeniu przypadku, niewydolność nerek u biorców przeszczepu nerki otrzymujących terapię skojarzoną interferonem alfa i rybawiryną była częstsza niż oczekiwano na podstawie wcześniejszych doświadczeń ośrodka z biorcami przeszczepu nerki nieotrzymującymi terapii skojarzonej. Związek niewydolności nerek z odrzuceniem przeszczepu nerki nie jest jasny.
Współzakażenie HIV lub HBV
Bezpieczeństwo i skuteczność PegIntron/REBETOL 200 mg i INTRON A/REBETOL w leczeniu pacjentów z HCV jednocześnie zakażonych HIV lub HBV nie zostały ustalone.
PRZEDAWKOWAĆ
Doświadczenie z przedawkowaniem jest ograniczone. Zgłaszano przypadki ostrego spożycia do 20 g kapsułek REBETOL, spożycia do 120 milionów jednostek INTRON A oraz dawek podskórnych INTRON A do 10 razy większych od zalecanych. Podstawowymi obserwowanymi efektami są zwiększona częstość występowania i nasilenie działań niepożądanych związanych z terapeutycznym stosowaniem INTRON A i REBETOL. Jednak po podaniu pojedynczych podskórnych dawek INTRON A, przekraczających zalecenia dotyczące dawkowania, zgłaszano zaburzenia enzymów wątrobowych, niewydolność nerek, krwotok i zawał mięśnia sercowego.
Nie ma swoistego antidotum na przedawkowanie INTRON A lub REBETOL 200 mg, a hemodializa i dializa otrzewnowa nie są skuteczne w leczeniu przedawkowania tych środków.
PRZECIWWSKAZANIA
Terapia skojarzona REBETOL jest przeciwwskazana w:
- kobiety w ciąży. REBETOL 200 mg może spowodować uszkodzenie płodu w przypadku podania kobiecie w ciąży. REBETOL jest przeciwwskazany u kobiet w ciąży lub mogących zajść w ciążę. Jeśli REBETOL jest stosowany w okresie ciąży lub jeśli pacjentka zajdzie w ciążę podczas przyjmowania leku REBETOL 200 mg, pacjentkę należy poinformować o potencjalnym zagrożeniu dla płodu [patrz OSTRZEŻENIA I ŚRODKI , Stosowanie w określonych populacjach , oraz INFORMACJA O PACJENCIE ]
- mężczyźni, których partnerki są w ciąży
- pacjenci ze znanymi reakcjami nadwrażliwości, takimi jak zespół Stevensa-Johnsona, toksyczne, martwicze oddzielanie się naskórka i rumień wielopostaciowy na rybawirynę lub którykolwiek składnik produktu
- pacjenci z autoimmunologicznym zapaleniem wątroby
- pacjenci z hemoglobinopatiami (np. talasemia major, anemia sierpowata)
- pacjenci z klirensem kreatyniny mniejszym niż 50 ml/min. [Widzieć Stosowanie w określonych populacjach oraz FARMAKOLOGIA KLINICZNA ]
- Jednoczesne podawanie produktu REBETOL i didanozyny jest przeciwwskazane, ponieważ zwiększa się ekspozycja na aktywny metabolit didanozyny (5'-trifosforan dideoksyadenozyny). U pacjentów otrzymujących didanozynę w skojarzeniu z rybawiryną opisywano niewydolność wątroby prowadzącą do zgonu, a także neuropatię obwodową, zapalenie trzustki i objawową hiperlaktatemię/kwasicę mleczanową [patrz INTERAKCJE Z LEKAMI ].
FARMAKOLOGIA KLINICZNA
Mechanizm akcji
Rybawiryna jest środkiem przeciwwirusowym [patrz Mikrobiologia ].
Farmakokinetyka
Właściwości farmakokinetyczne po podaniu pojedynczej i wielokrotnej dawki u dorosłych podsumowano w Tabeli 11. Po podaniu doustnym rybawiryna była szybko i intensywnie wchłaniana. Jednak ze względu na metabolizm pierwszego przejścia, bezwzględna biodostępność wynosiła średnio 64% (44%). Istniała liniowa zależność między dawką a AUCtf (AUC od czasu zero do ostatniego mierzalnego stężenia) po podaniu pojedynczych dawek od 200 do 1200 mg rybawiryny. Zależność między dawką a Cmax była krzywoliniowa, z tendencją do asymptoty powyżej pojedynczych dawek od 400 do 600 mg.
Po wielokrotnym podaniu doustnym, na podstawie AUC12hr, zaobserwowano 6-krotną kumulację rybawiryny w osoczu. Po podaniu doustnym dawki 600 mg dwa razy na dobę, stan stacjonarny osiągnięto po około 4 tygodniach, ze średnimi stężeniami w osoczu w stanie stacjonarnym wynoszącymi 2200 ng/ml (37%). Po przerwaniu dawkowania średni okres półtrwania wynosił 298 (30%) godzin, co prawdopodobnie odzwierciedla powolną eliminację z kompartmentów nieosoczowych.
Wpływ leków zobojętniających kwas na wchłanianie rybawiryny
Jednoczesne podawanie kapsułek REBETOL z lekami zobojętniającymi kwas zawierającymi magnez, glin i symetykon spowodowało zmniejszenie średniego AUCtf rybawiryny o 14%. Kliniczne znaczenie wyników tego badania z pojedynczą dawką nie jest znane.
Dystrybucja tkanek
Transport rybawiryny do przedziałów pozaplazmowych został najdokładniej zbadany w krwinkach czerwonych i zidentyfikowano, że odbywa się głównie za pośrednictwem równoważnego transportera nukleozydów typu es. Ten rodzaj transportera jest obecny praktycznie we wszystkich typach komórek i może odpowiadać za rozległą objętość dystrybucji. Rybawiryna nie wiąże się z białkami osocza.
Metabolizm i wydalanie
Rybawiryna ma dwa szlaki metabolizmu: (i) odwracalny szlak fosforylacji w komórkach jądrzastych; oraz (ii) szlak degradacji obejmujący derybozylację i hydrolizę amidu z wytworzeniem metabolitu kwasu triazolokarboksylowego. Rybawiryna i jej metabolity triazolokarboksamid i triazolo-kwas karboksylowy są wydalane przez nerki. Po podaniu doustnym 600 mg 14C-rybawiryny około 61% i 12% radioaktywności zostało wydalone odpowiednio z moczem i kałem w ciągu 336 godzin. Niezmieniona rybawiryna stanowiła 17% podanej dawki.
Specjalne populacje
Niewydolność nerek
Farmakokinetykę rybawiryny oceniano po podaniu pojedynczej dawki doustnej (400 mg) rybawiryny osobom niezakażonym HCV z różnym stopniem dysfunkcji nerek. Średnia wartość AUCtf była trzykrotnie większa u osób z wartościami klirensu kreatyniny od 10 do 30 ml/min w porównaniu z osobami z grupy kontrolnej (klirens kreatyniny większy niż 90 ml/min). U osób z wartościami klirensu kreatyniny od 30 do 60 ml/min, AUCtf było dwukrotnie większe w porównaniu z osobami kontrolnymi. Zwiększone AUCtf wydaje się być spowodowane zmniejszeniem klirensu nerkowego i pozanerkowego u tych osób. Badania skuteczności III fazy obejmowały osoby z wartościami klirensu kreatyniny powyżej 50 ml/min. Nie można dokładnie przewidzieć farmakokinetyki wielokrotnych dawek rybawiryny u pacjentów z zaburzeniami czynności nerek. Rybawiryna nie jest skutecznie usuwana przez hemodializę.
Pacjenci z klirensem kreatyniny mniejszym niż 50 ml/min nie powinni być leczeni produktem REBETOL [patrz PRZECIWWSKAZANIA ].
Zaburzenia czynności wątroby
Wpływ zaburzeń czynności wątroby oceniano po pojedynczej doustnej dawce rybawiryny (600 mg). Średnie wartości AUCtf nie różniły się istotnie u osób z łagodnymi, umiarkowanymi lub ciężkimi zaburzeniami czynności wątroby (klasyfikacja Child-Pugh A, B lub C) w porównaniu z osobami z grupy kontrolnej. Jednak średnie wartości Cmax wzrastały wraz z nasileniem zaburzeń czynności wątroby i były dwukrotnie większe u osób z ciężkimi zaburzeniami czynności wątroby w porównaniu z osobami z grupy kontrolnej.
Starsi pacjenci
Nie przeprowadzono oceny farmakokinetyki u osób w podeszłym wieku.
Płeć
W badaniu z pojedynczą dawką 18 mężczyzn i 18 kobiet nie stwierdzono klinicznie istotnych różnic w farmakokinetyce.
Pacjenci pediatryczni
Właściwości farmakokinetyczne wielokrotnych dawek preparatu REBETOL 200 mg w kapsułkach i INTRON A u dzieci z przewlekłym wirusowym zapaleniem wątroby typu C w wieku od 5 do 16 lat podsumowano w Tabeli 12. Farmakokinetyka REBETOL 200 mg i INTRON A (dawka znormalizowana) jest podobna u dorosłych i tematy pediatryczne.
Pełna charakterystyka farmakokinetyczna roztworu doustnego REBETOL nie została określona u dzieci. Wartości Cmin rybawiryny były podobne po podaniu preparatu REBETOL roztwór doustny lub REBETOL 200 mg kapsułki podczas 48 tygodni leczenia u dzieci (w wieku od 3 do 16 lat).
Przeprowadzono badanie kliniczne z udziałem dzieci z przewlekłym wirusowym zapaleniem wątroby typu C w wieku od 3 do 17 lat, w którym oceniano farmakokinetykę PegIntronu i REBETOL (kapsułki i roztwór doustny). U dzieci i młodzieży otrzymujących produkt PegIntron w dawce 60 μg/m²/tydzień, w dawce dostosowanej do powierzchni ciała, szacowany współczynnik transformacji logarytmicznej ekspozycji w okresie pomiędzy kolejnymi dawkami był o 58% [90% CI: 141%, 177%] wyższy niż obserwowane u dorosłych otrzymujących 1,5 mcg/kg/tydzień. Farmakokinetyka REBETOL-u (znormalizowana dawka) w tym badaniu była podobna do tej opisanej we wcześniejszym badaniu REBETOL 200 mg w skojarzeniu z INTRON A u dzieci i dorosłych.
Wpływ pokarmu na wchłanianie rybawiryny
Zarówno AUCtf, jak i Cmax wzrosły o 70%, gdy kapsułki REBETOL podawano z posiłkiem wysokotłuszczowym (841 kcal, 53,8 g tłuszczu, 31,6 g białka i 57,4 g węglowodanów) w badaniu farmakokinetyki pojedynczej dawki [patrz DAWKOWANIE I SPOSÓB PODAWANIA ].
Mikrobiologia
Mechanizm akcji
Mechanizm, dzięki któremu rybawiryna przyczynia się do jej skuteczności przeciwwirusowej w klinice, nie jest w pełni poznany. Rybawiryna ma bezpośrednie działanie przeciwwirusowe w hodowli tkankowej przeciwko wielu wirusom RNA. Rybawiryna zwiększa częstość mutacji w genomach kilku wirusów, a trifosforan rybawiryny hamuje polimerazę HCV w reakcji biochemicznej.
Aktywność przeciwwirusowa w hodowli komórkowej
Działanie przeciwwirusowe rybawiryny w replikonie HCV nie jest dobrze poznane i nie zostało zdefiniowane ze względu na toksyczność komórkową rybawiryny. Bezpośrednie działanie przeciwwirusowe zaobserwowano w kulturach tkankowych innych wirusów RNA. Aktywność interferonu przeciwko HCV wykazano w komórce zawierającej samoreplikujące się HCV-RNS (komórki replikonowe HCV) lub zakażenie HCV.
Opór
Genotypy HCV wykazują dużą zmienność w ich odpowiedzi na terapię pegylowanym rekombinowanym ludzkim interferonem/rybawiryną. Zmiany genetyczne związane ze zmienną odpowiedzią nie zostały zidentyfikowane.
Opór krzyżowy
Nie ma doniesień o oporności krzyżowej między pegylowanymi/niepegylowanymi interferonami a rybawiryną.
Toksykologia i farmakologia zwierząt
Długoterminowe badania na myszach i szczurach [18 do 24 miesięcy; dawki odpowiednio 20 do 75 i 10 do 40 mg/kg/dobę (szacunkowe dawki ekwiwalentne dla ludzi wynoszą odpowiednio 1,67 do 6,25 i 1,43 do 5,71 mg/kg/dobę, na podstawie dostosowania powierzchni ciała dla osoby dorosłej o masie 60 kg; około 0,1 do 0,4 razy większa od maksymalnej 24-godzinnej dawki rybawiryny u ludzi)] wykazali związek między przewlekłą ekspozycją na rybawirynę a zwiększoną częstością występowania zmian naczyniowych (krwotoków mikroskopowych) u myszy. U szczurów zwyrodnienie siatkówki wystąpiło w grupie kontrolnej, ale częstość występowania była zwiększona u szczurów leczonych rybawiryną.
badaniu, w którym młode szczury otrzymywały po urodzeniu rybawirynę w dawkach 10, 25 i 50 mg/kg/dobę, zgony związane z lekiem wystąpiły przy dawce 50 mg/kg (przy stężeniach młodych szczurów w osoczu poniżej stężeń w osoczu u ludzi dawka terapeutyczna) między dniem 13. a 48. badania. Szczenięta szczurów, którym podawano dawki od 7. do 63. dnia po urodzeniu, wykazywały niewielkie, zależne od dawki zmniejszenie ogólnego wzrostu we wszystkich dawkach, które następnie objawiało się niewielkim spadkiem masy ciała, długości ciemieniowo-zadu, i długość kości. Efekty te wykazały dowody na odwracalność i nie zaobserwowano żadnego wpływu histopatologicznego na kości. Nie zaobserwowano wpływu rybawiryny na rozwój neurobehawioralny lub reprodukcyjny.
Studia kliniczne
W badaniu klinicznym 1 oceniano monoterapię PegIntronem. Widzieć Oznakowanie PegIntron w celu uzyskania informacji o tym badaniu.
Terapia skojarzona REBETOL/PegIntron
Osoby dorosłe
Studium 2
Randomizowane badanie porównało leczenie z dwoma schematami PegIntron/REBETOL 200 mg [PegIntron 1,5 µg/kg podskórnie raz w tygodniu/REBETOL 800 mg doustnie dziennie (w dawkach podzielonych); PegIntron 1,5 µg/kg podskórnie raz w tygodniu przez 4 tygodnie, następnie 0,5 µg/kg podskórnie raz w tygodniu przez 44 tygodnie/REBETOL 1000 lub 1200 mg doustnie dziennie (w dawkach podzielonych)] z INTRON A [3 mln jm podskórnie trzy razy w tygodniu/REBETOL 1000 lub 1200 mg doustnie na dobę (w dawkach podzielonych)] u 1530 osób dorosłych z przewlekłym wirusowym zapaleniem wątroby typu C. Osoby nieleczone wcześniej interferonem były leczone przez 48 tygodni i obserwowane przez 24 tygodnie po leczeniu. Zakwalifikowani pacjenci mieli wyrównaną chorobę wątroby, wykrywalny HCV-RNA, podwyższoną aktywność ALT i histopatologię wątroby zgodną z przewlekłym zapaleniem wątroby.
Odpowiedź na leczenie zdefiniowano jako niewykrywalny HCV-RNA 24 tygodnie po leczeniu (patrz Tabela 13). Wskaźnik odpowiedzi na PegIntron 1,5 μg/kg i rybawirynę w dawce 800 mg był wyższy niż wskaźnik odpowiedzi na INTRON A/REBETOL (patrz Tabela 13). Wskaźnik odpowiedzi na PegIntron 1,5 → 0,5 μg/kg/REBETOL 200 mg był zasadniczo taki sam jako odpowiedź na INTRON A/REBETOL (dane nie pokazane).
Pacjenci z genotypem 1 wirusa, niezależnie od miana wirusa, mieli niższy wskaźnik odpowiedzi na PegIntron (1,5 µg/kg)/REBETOL (800 mg) w porównaniu z osobami z innymi genotypami wirusa. U pacjentów z obydwoma złymi czynnikami prognostycznymi (genotyp 1 i wysokie miano wirusa) odsetek odpowiedzi wynosił 30% (78/256) w porównaniu z odsetkiem odpowiedzi 29% (71/247) w przypadku terapii skojarzonej INTRON A/REBETOL.
Osoby z niższą masą ciała miały tendencję do częstszego występowania niepożądanych reakcji [patrz DZIAŁANIA NIEPOŻĄDANE i wyższe wskaźniki odpowiedzi niż osoby o większej masie ciała. Różnice w odsetku odpowiedzi między ramionami leczenia nie różniły się istotnie w zależności od masy ciała.
Wskaźniki odpowiedzi na leczenie skojarzone PegIntron/REBETOL wynosiły 49% u mężczyzn i 56% u kobiet. Wskaźniki odpowiedzi były niższe u Afroamerykanów i Latynosów i wyższe u Azjatów w porównaniu z osobami rasy białej. Chociaż Afroamerykanie mieli wyższy odsetek słabych czynników prognostycznych w porównaniu z osobami rasy kaukaskiej, liczba badanych osób niekaukaskich (11% wszystkich) była niewystarczająca, aby umożliwić wyciągnięcie znaczących wniosków na temat różnic w odsetkach odpowiedzi po dostosowaniu do czynników prognostycznych w tym badaniu.
Biopsje wątroby wykonano przed i po leczeniu u 68% badanych. W porównaniu ze stanem wyjściowym u około dwóch trzecich pacjentów we wszystkich grupach leczenia zaobserwowano niewielkie zmniejszenie stanu zapalnego.
Studium 3
dużym badaniu pozaszpitalnym w Stanach Zjednoczonych 4913 pacjentów z przewlekłym wirusowym zapaleniem wątroby typu C zostało losowo przydzielonych do grupy otrzymującej PegIntron 1,5 µg/kg podskórnie raz w tygodniu w skojarzeniu z dawką REBETOL 200 mg od 800 do 1400 mg (dawkowanie zależne od masy ciała [WBD]) lub 800 mg (na płasko) doustnie dziennie (w dawkach podzielonych) przez 24 lub 48 tygodni w zależności od genotypu. Odpowiedź na leczenie została zdefiniowana jako niewykrywalny HCV-RNA (na podstawie testu z dolną granicą wykrywalności 125 IU/ml) 24 tygodnie po leczeniu.
Leczenie produktem PegIntron 1,5 mcg/kg i REBETOL 800 do 1400 mg skutkowało wyższą trwałą odpowiedzią wirusologiczną w porównaniu do PegIntron w połączeniu ze stałą dawką dobową REBETOL 800 mg. Osoby o masie ciała powyżej 105 kg odniosły największą korzyść z WBD, chociaż niewielką korzyść zaobserwowano również u osób o masie ciała powyżej 85 do 105 kg (patrz Tabela 14). Korzyści z WBD u osób ważących powyżej 85 kg zaobserwowano w przypadku genotypów HCV 1-3. Nie było wystarczających danych, aby wyciągnąć wnioski dotyczące innych genotypów. Stosowanie WBD powodowało zwiększoną częstość występowania anemii [patrz DZIAŁANIA NIEPOŻĄDANE ].
Łącznie 1552 pacjentów o masie ciała powyżej 65 kg w badaniu 3 miało genotyp 2 lub 3 i zostało losowo przydzielonych do 24 lub 48 tygodni terapii. Nie zaobserwowano dodatkowych korzyści przy dłuższym czasie leczenia.
Studium 4
dużym randomizowanym badaniu porównano bezpieczeństwo i skuteczność leczenia przez 48 tygodni z dwoma schematami PegIntron/REBETOL 200 mg [PegIntron 1,5 µg/kg i 1 µg/kg podskórnie raz dawki)] i Pegasys 180 µg podskórnie raz w tygodniu w skojarzeniu z Copegus 1000 do 1200 mg PO na dobę (w dwóch dawkach podzielonych) u 3070 nieleczonych dorosłych z przewlekłym wirusowym zapaleniem wątroby typu C o genotypie 1. W tym badaniu brak wczesnej odpowiedzi wirusologicznej (niewykrywalny HCV-RNA lub większe lub równe 2 log10 redukcji w stosunku do wartości wyjściowych) w 12. tygodniu leczenia było kryterium przerwania leczenia. SVR zdefiniowano jako niewykrywalny HCV-RNA (test Roche COBAS TaqMan, dolna granica oznaczalności 27 j.m./ml) w 24 tygodnie po leczeniu (patrz Tabela 15).
Ogólne wskaźniki SVR były podobne w trzech grupach leczenia. Niezależnie od grupy leczenia, wskaźniki SVR były niższe u osób ze złymi czynnikami prognostycznymi. Pacjenci ze słabymi czynnikami prognostycznymi, przydzieleni losowo do grupy PegIntron (1,5 µg/kg)/REBETOL lub Pegasys/Copegus, osiągnęli jednak wyższe wskaźniki SVR w porównaniu z podobnymi osobami przydzielonymi losowo do grupy PegIntron 1 µg/kg/REBETOL. Dla dawki PegIntron 1,5 mcg/kg i REBETOL, wskaźniki SVR u pacjentów z następującymi czynnikami prognostycznymi i bez nich były następujące: marskość wątroby (10% vs. 42%), normalna aktywność AlAT (32% vs. 42%), początkowa wirusowość obciążenie większe niż 600 000 IU/ml (35% wobec 61%), osoby w wieku 40 lat i starsze (38% wobec 50%) oraz rasa afroamerykańska (23% wobec 44%). U pacjentów z niewykrywalnym HCV-RNA w 12. tygodniu leczenia, którzy otrzymywali PegIntron (1,5 µg/kg)/REBETOL 200 mg, odsetek SVR wyniósł 81% (328/407).
Badanie 5 – Terapia skojarzona REBETOL/PegIntron w przypadku niepowodzeń wcześniejszego leczenia
badaniu nieporównawczym 2293 pacjentów z umiarkowanym lub ciężkim zwłóknieniem, u których wcześniejsze leczenie skojarzeniem interferonu alfa z rybawiryną nie powiodło się, ponownie leczono preparatem PegIntron w dawce 1,5 mcg/kg podskórnie raz w tygodniu w skojarzeniu z rybawiryną dostosowaną do masy ciała. Kwalifikujące się osoby obejmowały wcześniej nieodpowiadające na leczenie (pacjenci, którzy byli HCV-RNA dodatni pod koniec co najmniej 12 tygodni leczenia) i wcześniejszy nawrót (pacjenci, którzy mieli ujemny wynik HCVRNA pod koniec co najmniej 12 tygodni leczenia, a następnie mieli nawrót choroby po zakończeniu leczenia podejmować właściwe kroki). Osobnicy, którzy mieli wynik ujemny w 12. tygodniu, byli leczeni przez 48 tygodni i obserwowani przez 24 tygodnie po leczeniu. Odpowiedź na leczenie zdefiniowano jako niewykrywalny HCV-RNA w 24 tygodnie po leczeniu (mierzony testem opartym na badaniach, granica wykrywalności 125 IU/ml). Całkowity odsetek odpowiedzi wyniósł 22% (497/2293) (99% CI: 19,5; 23,9). Pacjenci z następującymi cechami mieli mniejsze szanse na odniesienie korzyści z ponownego leczenia: poprzedni brak odpowiedzi, wcześniejsze leczenie pegylowanym interferonem, znaczące zwłóknienie mostkowe lub marskość wątroby oraz zakażenie genotypu 1.
Wskaźniki utrzymującej się odpowiedzi wirusologicznej na ponowne leczenie według charakterystyki wyjściowej podsumowano w Tabeli 16.
Osiągnięcie niewykrywalnego HCV-RNA w 12. tygodniu leczenia było silnym predyktorem SVR. W tym badaniu 1470 (64%) pacjentów nie osiągnęło niewykrywalnego HCV-RNA w 12. tygodniu leczenia i zaproponowano im włączenie do badań długoterminowych z powodu niewystarczającej odpowiedzi na leczenie. Spośród 823 (36%) pacjentów, u których RNA HCV było niewykrywalne w 12. tygodniu leczenia, osoby zakażone genotypem 1 miały SVR wynoszące 48% (245/507), z zakresem odpowiedzi według skali zwłóknienia (F4-F2) wynoszącym 39-55%. Pacjenci zakażeni genotypem 2/3, u których RNA HCV były niewykrywalne w 12. tygodniu leczenia, mieli ogólną SVR wynoszącą 70% (196/281), z zakresem odpowiedzi na podstawie punktacji zwłóknienia (F4-F2) wynoszącym 60-83%. W przypadku wszystkich genotypów wyższe wyniki zwłóknienia wiązały się ze zmniejszonym prawdopodobieństwem osiągnięcia SVR.
Pacjenci pediatryczni
Wcześniej nieleczeni pacjenci pediatryczni w wieku od 3 do 17 lat z wyrównanym przewlekłym wirusowym zapaleniem wątroby typu C i wykrywalnym HCV-RNA otrzymywali REBETOL 15 mg/kg na dobę i PegIntron 60 μg/m2 raz w tygodniu przez 24 lub 48 tygodni w oparciu o genotyp HCV i początkowe wartości wirusa Załaduj. Wszystkie osoby miały być obserwowane przez 24 tygodnie po leczeniu. W sumie 107 pacjentów otrzymało leczenie, z czego 52% stanowiły kobiety, 89% było rasy kaukaskiej, a 67% było zakażonych genotypem 1 HCV. Osoby zakażone genotypem 1, 4 lub genotypem 3 z HCV-RNA większym lub równym 600 000 IU/ml otrzymały 48 tygodni terapii, podczas gdy osoby zakażone Genotypem 2 lub Genotypem 3 z HCV-RNA mniejszym niż 600 000 IU/ml otrzymały 24 tygodnie terapii. Wyniki badań podsumowano w Tabeli 17.
Terapia skojarzona REBETOL/INTRON A
Osoby dorosłe
Osoby wcześniej nieleczone
Dorośli z wyrównanym przewlekłym wirusowym zapaleniem wątroby typu C i wykrywalnym HCV-RNA (ocenianym przez laboratorium centralne przy użyciu opartego na badaniach testu RT-PCR), którzy wcześniej nie byli leczeni interferonem alfa, zostali zakwalifikowani do dwóch wieloośrodkowych, podwójnie ślepych badań (amerykańskich i międzynarodowych). i przydzieleni losowo do otrzymywania kapsułek REBETOL 200 mg 1200 mg/dobę (1000 mg/dobę dla osób o masie ciała mniejszej lub równej 75 kg) i INTRON A 3 mln jm trzy razy w tygodniu lub INTRON A i placebo przez 24 lub 48 tygodni, a następnie 24 tygodnie obserwacja poza terapią. Międzynarodowe badanie nie obejmowało 24-tygodniowego ramienia leczenia INTRON A i placebo. Do badania w USA włączono 912 pacjentów, którzy na początku badania stanowili 67% mężczyzn, 89% rasy kaukaskiej, ze średnią punktacją Knodella HAI (I+II+III) wynoszącą 7,5 i 72% z genotypem 1. Międzynarodowe badanie przeprowadzone w Europie, Izraelu , Kanada i Australia, do badania włączono 799 pacjentów (65% mężczyzn, 95% rasy kaukaskiej, średni wynik w skali Knodella 6,8 i 58% z genotypem 1).
Wyniki badań podsumowano w Tabeli 18.
Spośród pacjentów, którzy nie uzyskali HCV-RNA poniżej granicy wykrywalności testu opartego na badaniach do 24. tygodnia leczenia REBETOL/INTRON A, mniej niż 5% odpowiedziało na dodatkowe 24 tygodnie leczenia skojarzonego.
Wśród pacjentów z genotypem 1 HCV leczonych terapią REBETOL/INTRON A, którzy osiągnęli HCV-RNA poniżej granicy wykrywalności testu opartego na badaniach do 24. tygodnia, osoby przydzielone losowo do 48 tygodni leczenia miały wyższą odpowiedź wirusologiczną w porównaniu z osobami w grupie 24-tygodniowej. grupa tygodniowa. Nie zaobserwowano wzrostu odsetka odpowiedzi u pacjentów z HCV bez genotypu 1, przydzielonych losowo do terapii REBETOL/INTRON A przez 48 tygodni w porównaniu z 24 tygodniami.
Tematy nawrotu
Pacjenci z skompensowanym przewlekłym wirusowym zapaleniem wątroby typu C i wykrywalnym HCV-RNA (ocenianym przez laboratorium centralne przy użyciu opartego na badaniach testu RT-PCR), u których doszło do nawrotu po jednym lub dwóch cyklach leczenia interferonem (zdefiniowanym jako nieprawidłowe poziomy ALT w surowicy), zostali zakwalifikowani do dwóch wieloośrodkowe, podwójnie zaślepione badania (amerykańskie i międzynarodowe) i randomizowane do otrzymywania REBETOL 1200 mg/dobę (1000 mg/dobę dla osób o masie ciała ≤ 75 kg) i INTRON A 3 mln IU trzy razy w tygodniu lub INTRON A i placebo przez 24 tygodnie, a następnie 24 tygodnie obserwacji poza terapią. Do badania w USA włączono 153 pacjentów, którzy na początku stanowili 67% mężczyzn, 92% rasy kaukaskiej, ze średnią punktacją Knodella HAI (I+II+III) wynoszącą 6,8 i 58% z genotypem 1. Międzynarodowe badanie przeprowadzone w Europie, Izraelu , Kanada i Australia, do badania włączono 192 osoby (64% mężczyzn, 95% rasy kaukaskiej, średni wynik w skali Knodella 6,6 i 56% genotyp 1). Wyniki badań podsumowano w Tabeli 19.
Odpowiedzi wirusologiczne i histologiczne były podobne u mężczyzn i kobiet zarówno w badaniach wcześniej nieleczonych, jak iw badaniach z nawrotami.
Pacjenci pediatryczni
Dzieci w wieku od 3 do 16 lat z wyrównanym przewlekłym wirusowym zapaleniem wątroby typu C i wykrywalnym HCV-RNA (ocenianym przez laboratorium centralne przy użyciu testu RT-PCR opartego na badaniach) leczono preparatem REBETOL 15 mg/kg na dobę i INTRON A 3 mln j.m./ m² trzy razy w tygodniu przez 48 tygodni, a następnie 24 tygodnie po zakończeniu terapii. Łącznie 118 pacjentów zostało poddanych leczeniu, z czego 57% stanowili mężczyźni, 80% rasy kaukaskiej, a 78% osoby o genotypie 1. Osoby w wieku poniżej 5 lat otrzymywały roztwór doustny REBETOL, a osoby w wieku 5 lat lub starsze otrzymywały roztwór doustny REBETOL lub kapsułki.
Wyniki badań podsumowano w Tabeli 20.
Pacjenci z genotypem 1 wirusa, niezależnie od miana wirusa, mieli niższy wskaźnik odpowiedzi na terapię skojarzoną INTRON A/REBETOL w porównaniu do pacjentów z genotypem nie-1, 36% w porównaniu z 81%. U osób z obydwoma złymi czynnikami prognostycznymi (genotyp 1 i wysokie miano wirusa) odsetek odpowiedzi wyniósł 26% (13/50).
INFORMACJA O PACJENCIE
Brak informacji. Proszę odnieść się do OSTRZEŻENIA I ŚRODKI Sekcja.