Voltaren 100mg, 50mg Diclofenac Zastosowanie, skutki uboczne i dawkowanie. Cena w aptece internetowej. Leki generyczne bez recepty.
Co to jest Voltaren i jak się go stosuje?
Voltaren stosuje się w leczeniu objawów bólu związanego z reumatoidalnym zapaleniem stawów, chorobą zwyrodnieniową stawów, zesztywniającym zapaleniem stawów kręgosłupa, bolesnym miesiączkowaniem oraz bólem o nasileniu od łagodnego do umiarkowanego. Voltaren 100mg może być stosowany samodzielnie lub z innymi lekami.
Voltaren należy do klasy leków zwanych niesteroidowymi lekami przeciwzapalnymi (NLPZ).
Jakie są możliwe skutki uboczne Voltaren 50mg?
Voltaren może powodować poważne skutki uboczne, w tym:
- ból głowy,
- głód,
- wyzysk,
- drażliwość,
- zawroty głowy,
- mdłości,
- szybkie tętno,
- uczucie niepokoju lub roztrzęsienia,
- drętwienie lub mrowienie dłoni, ramion, nóg lub stóp,
- osłabienie ramion, rąk, nóg lub stóp,
- piekący ból ramion, dłoni, nóg lub stóp,
- poważne zmiany nastroju lub zachowania,
- nerwowość,
- dezorientacja,
- podniecenie,
- paranoja,
- halucynacje,
- problemy z pamięcią,
- problemy z koncentracją,
- myśli samobójcze,
- zerwanie ścięgna,
- nagły ból,
- obrzęk,
- siniaki,
- czułość,
- sztywność,
- problemy ruchowe,
- dźwięk trzaskania lub trzaskania w dowolnym stawie,
- silny ból brzucha,
- biegunka wodnista lub krwawa,
- fruwające w piersi,
- duszność,
- wysypka na skórze,
- problemy z oddychaniem,
- drgawki (konwulsje),
- silne bóle głowy,
- Problemy ze wzrokiem,
- ból za oczami,
- ból w górnej części brzucha,
- utrata apetytu,
- ciemny mocz,
- taborety w kolorze gliny i
- zażółcenie skóry lub oczu (żółtaczka)
i natychmiast uzyskać pomoc medyczną, jeśli wystąpi którykolwiek z wymienionych powyżej objawów.
Najczęstsze skutki uboczne Voltaren to:
- niestrawność,
- gaz,
- ból brzucha,
- mdłości,
- wymioty,
- biegunka,
- zaparcie,
- ból głowy,
- zawroty głowy,
- senność,
- zatkany nos,
- swędzący,
- zwiększona potliwość,
- podwyższone ciśnienie krwi i
- obrzęk lub ból rąk lub nóg
Poinformuj lekarza, jeśli wystąpią jakiekolwiek skutki uboczne, które Ci przeszkadzają lub które nie ustępują.
To nie wszystkie możliwe skutki uboczne Voltarena. Aby uzyskać więcej informacji, skontaktuj się z lekarzem lub farmaceutą.
Zadzwoń do lekarza, aby uzyskać poradę medyczną na temat skutków ubocznych. Możesz zgłosić skutki uboczne do FDA pod numerem 1-800-FDA-1088.
OSTRZEŻENIE
TOKSYCZNOŚĆ HEPATOTOKSYCZNOŚĆ, TOKSYCZNOŚĆ SERCA, TOKSYCZNOŚĆ ZARODKO-PŁODOWA
Zdarzenia zakrzepowo-sercowe
- Niesteroidowe leki przeciwzapalne (NLPZ) zwiększają ryzyko poważnych incydentów zakrzepowych układu krążenia, w tym zawału mięśnia sercowego i udaru mózgu, które mogą być śmiertelne. Ryzyko to może wystąpić na początku leczenia i może nasilać się wraz z czasem stosowania (patrz OSTRZEŻENIA).
- VOLTAREN® jest przeciwwskazany w przypadku operacji pomostowania aortalno-wieńcowego (CABG) (patrz PRZECIWWSKAZANIA, OSTRZEŻENIA).
Krwawienie z przewodu pokarmowego, owrzodzenie i perforacja
- NLPZ powodują zwiększone ryzyko poważnych działań niepożądanych ze strony przewodu pokarmowego (GI), w tym krwawienia, owrzodzenia i perforacji żołądka lub jelit, które mogą być śmiertelne. Zdarzenia te mogą wystąpić w dowolnym momencie podczas użytkowania i bez objawów ostrzegawczych. Pacjenci w podeszłym wieku oraz pacjenci z chorobą wrzodową i/lub krwawieniem z przewodu pokarmowego w wywiadzie są bardziej narażeni na poważne zdarzenia żołądkowo-jelitowe. (patrz OSTRZEŻENIA).
OPIS
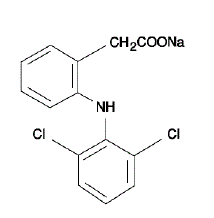
VOLTAREN® (tabletki dojelitowe diklofenaku sodu) jest pochodną kwasu benzenooctowego. VOLTAREN 50 mg jest dostępny w postaci tabletek o opóźnionym uwalnianiu (powlekanych dojelitowo) po 75 mg (jasnoróżowy) do podawania doustnego. Diklofenak sodowy jest białym lub lekko żółtawym krystalicznym proszkiem i jest słabo rozpuszczalny w wodzie w temperaturze 25°C. Nazwa chemiczna to sól monosodowa kwasu 2-[(2,6-dichlorofenylo)amino]benzenooctowego. Masa cząsteczkowa wynosi 318,14. Jego wzór cząsteczkowy to C14H10Cl2NNaO2 i ma następujący wzór strukturalny:
Składniki nieaktywne leku VOLTAREN to: hydroksypropylometyloceluloza, tlenek żelaza, laktoza, stearynian magnezu, kopolimer kwasu metakrylowego, celuloza mikrokrystaliczna, glikol polietylenowy, powidon, glikol propylenowy, wodorotlenek sodu, glikolan sodowy skrobi, talk, dwutlenek tytanu.
WSKAZANIA
Przed podjęciem decyzji o zastosowaniu leku VOLTAREN należy dokładnie rozważyć potencjalne korzyści i ryzyko związane ze stosowaniem leku VOLTAREN® (tabletki dojelitowe diklofenak sodowy) oraz innych opcji leczenia. Należy stosować najmniejszą skuteczną dawkę przez najkrótszy czas, zgodnie z indywidualnymi celami leczenia pacjenta (patrz OSTRZEŻENIA ; Krwawienie z przewodu pokarmowego , Owrzodzenie , oraz Perforacja ).
VOLTAREN jest wskazany:
- do łagodzenia oznak i objawów choroby zwyrodnieniowej stawów
- do łagodzenia oznak i objawów reumatoidalnego zapalenia stawów
- do ostrego lub długotrwałego stosowania w łagodzeniu oznak i objawów zesztywniającego zapalenia stawów kręgosłupa
DAWKOWANIE I SPOSÓB PODAWANIA
Przed podjęciem decyzji o zastosowaniu leku VOLTAREN należy dokładnie rozważyć potencjalne korzyści i ryzyko związane ze stosowaniem leku VOLTAREN® (tabletki dojelitowe diklofenak sodowy) oraz innych opcji leczenia. Należy stosować najmniejszą skuteczną dawkę przez najkrótszy czas, zgodnie z indywidualnymi celami leczenia pacjenta (patrz OSTRZEŻENIA ; Krwawienie z przewodu pokarmowego, owrzodzenie i perforacja ).
Po zaobserwowaniu odpowiedzi na początkowe leczenie preparatem VOLTAREN 100 mg, dawkę i częstotliwość należy dostosować do indywidualnych potrzeb pacjenta.
W celu złagodzenia choroby zwyrodnieniowej stawów zalecana dawka wynosi od 100 do 150 mg na dobę w dawkach podzielonych (50 mg dwa razy na dobę lub trzy razy na dobę lub 75 mg dwa razy na dobę).
celu złagodzenia reumatoidalnego zapalenia stawów zalecana dawka wynosi 150 do 200 mg/dobę w dawkach podzielonych (50 mg trzy razy na dobę lub cztery razy na dobę lub 75 mg dwa razy na dobę).
W celu złagodzenia zesztywniającego zapalenia stawów kręgosłupa zalecana dawka wynosi od 100 do 125 mg/dobę, podawana jako 25 mg cztery razy na dobę, z dodatkową dawką 25 mg przed snem, jeśli to konieczne.
Różne preparaty diklofenaku [VOLTAREN® (tabletki dojelitowe diklofenaku sodu); VOLTAREN®®XR (tabletki diklofenaku sodu o przedłużonym uwalnianiu); CATAFLAM® (tabletki o natychmiastowym uwalnianiu diklofenaku potasu)] niekoniecznie są biorównoważne, nawet jeśli moc miligramów jest taka sama.
JAK DOSTARCZONE
VOLTAREN® (tabletki dojelitowe diklofenak sodowy) - 75 mg
Jasnoróżowe, obustronnie wypukłe, trójkątne tabletki dojelitowe (nadruk „VOLTAREN 75” po jednej stronie czarnym tuszem)
Butelki 100 NDC 0028-0264-01
Przechowywać w temperaturze pokojowej 20°C do 25°C (68°F do 77°F); wycieczki dozwolone między 15°C a 30°C (59°F i 86°F) [patrz Temperatura pokojowa kontrolowana przez USP ].
Chronić przed wilgocią. Dozować w szczelnym pojemniku (USP).
Dystrybutor: Novartis Pharmaceuticals Corporation East Hanover, NJ 07936. Aktualizacja: kwiecień 2021
SKUTKI UBOCZNE
Następujące działania niepożądane omówiono bardziej szczegółowo w innych punktach oznakowania:
- Sercowo-naczyniowe zdarzenia zakrzepowe (patrz OSTRZEŻENIA )
- Krwawienie, owrzodzenie i perforacja przewodu pokarmowego (patrz OSTRZEŻENIA )
- Hepatotoksyczność (patrz OSTRZEŻENIA )
- Nadciśnienie (patrz OSTRZEŻENIA )
- Niewydolność serca i obrzęk (patrz OSTRZEŻENIA )
- Toksyczność nerek i hiperkaliemia (patrz OSTRZEŻENIA )
- Reakcje anafilaktyczne (patrz OSTRZEŻENIA )
- Poważne reakcje skórne (patrz OSTRZEŻENIA )
- Toksyczność hematologiczna (patrz OSTRZEŻENIA )
Doświadczenie w badaniach klinicznych
Ponieważ badania kliniczne prowadzone są w bardzo zróżnicowanych warunkach, częstość występowania działań niepożądanych obserwowanych w badaniach klinicznych leku nie może być bezpośrednio porównywana z częstością w badaniach klinicznych innego leku i może nie odzwierciedlać częstości obserwowanych w praktyce.
U pacjentów przyjmujących VOLTAREN® (tabletki dojelitowe diklofenak sodowy) lub inne NLPZ, najczęściej zgłaszanymi działaniami niepożądanymi występującymi u około 1% do 10% pacjentów są: Doświadczenia żołądkowo-jelitowe, w tym: ból brzucha, zaparcia, biegunka, niestrawność, wzdęcia, duże krwawienie/perforacja, zgaga, nudności, wrzody przewodu pokarmowego (żołądka/dwunastnicy) i wymioty.
Nieprawidłowa czynność nerek, niedokrwistość, zawroty głowy, obrzęki, podwyższona aktywność enzymów wątrobowych, bóle głowy, wydłużony czas krwawienia, świąd, wysypki i szumy uszne. Dodatkowe zdarzenia niepożądane zgłaszane sporadycznie obejmują:
Ciało jako całość: gorączka, infekcja, posocznica
Układu sercowo-naczyniowego: zastoinowa niewydolność serca, nadciśnienie, tachykardia, omdlenie
Układ trawienny: suchość błony śluzowej jamy ustnej, zapalenie przełyku, wrzody żołądka/wrzodów trawiennych, zapalenie żołądka, krwawienie z przewodu pokarmowego, zapalenie języka, krwawe wymioty, zapalenie wątroby, żółtaczka
Układ hemiczny i limfatyczny: wybroczyny, eozynofilia, leukopenia, melena, plamica, krwawienie z odbytu, zapalenie jamy ustnej, małopłytkowość
Metaboliczne i odżywcze: zmiany wagi
System nerwowy: lęk, osłabienie, splątanie, depresja, zaburzenia snów, senność, bezsenność, złe samopoczucie, nerwowość, parestezje, senność, drżenie, zawroty głowy
Układ oddechowy: astma, duszność
Skóra i przydatki: łysienie, nadwrażliwość na światło, zwiększona potliwość
Zmysły specjalne: rozmazany obraz
Układ moczowo-płciowy: zapalenie pęcherza moczowego, bolesne oddawanie moczu, krwiomocz, śródmiąższowe zapalenie nerek, skąpomocz/wielomocz, białkomocz, niewydolność nerek
Inne działania niepożądane, które występują rzadko to:
Ciało jako całość: reakcje anafilaktyczne, zmiany apetytu, śmierć
Układu sercowo-naczyniowego: arytmia, niedociśnienie, zawał mięśnia sercowego, kołatanie serca, zapalenie naczyń
Układ trawienny: zapalenie okrężnicy, odbijanie się, piorunujące zapalenie wątroby z żółtaczką lub bez, niewydolność wątroby, martwica wątroby, zapalenie trzustki
Układ hemiczny i limfatyczny: agranulocytoza, niedokrwistość hemolityczna, niedokrwistość aplastyczna, powiększenie węzłów chłonnych, pancytopenia
Metaboliczne i odżywcze: hiperglikemia
System nerwowy: drgawki, śpiączka, halucynacje, zapalenie opon mózgowych
Układ oddechowy: depresja oddechowa, zapalenie płuc
Skóra i przydatki: obrzęk naczynioruchowy, toksyczna nekroliza naskórka, rumień wielopostaciowy, złuszczające zapalenie skóry, zespół Stevensa-Johnsona, pokrzywka
Zmysły specjalne: zapalenie spojówek, upośledzenie słuchu
PRZEDAWKOWAĆ
Objawy występujące po ostrym przedawkowaniu NLPZ były zazwyczaj ograniczone do letargu, senności, nudności, wymiotów i bólu w nadbrzuszu, które na ogół ustępowały po zastosowaniu leczenia wspomagającego. Wystąpiło krwawienie z przewodu pokarmowego. Wystąpiło nadciśnienie, ostra niewydolność nerek, depresja oddechowa i śpiączka, ale występowały one rzadko (patrz OSTRZEŻENIA ; Sercowo-naczyniowe zdarzenia zakrzepowe, krwawienie z przewodu pokarmowego, owrzodzenie i nadciśnienie, toksyczność nerek i hiperkaliemia ).
Prowadź pacjentów z opieką objawową i podtrzymującą po przedawkowaniu NLPZ. Nie ma swoistych odtrutek. Rozważyć wymioty i/lub węgiel aktywowany (60 do 100 gramów u dorosłych, 1 do 2 gramów na kg masy ciała u dzieci) i/lub osmotyczne przeczyszczanie u pacjentów z objawami obserwowanymi w ciągu czterech godzin od spożycia lub u pacjentów z dużym przedawkowaniem ( 5 do 10 razy więcej niż zalecana dawka). Wymuszona diureza, alkalizacja moczu, hemodializa lub hemoperfuzja mogą nie być przydatne ze względu na wysokie wiązanie z białkami.
Aby uzyskać dodatkowe informacje na temat leczenia przedawkowania, skontaktuj się z centrum kontroli zatruć (1-800-222-1222).
INTERAKCJE Z LEKAMI
W Tabeli 2 przedstawiono klinicznie istotne interakcje leków z diklofenakiem.
OSTRZEŻENIA
Zdarzenia zakrzepowo-sercowe
Badania kliniczne kilku selektywnych i nieselektywnych NLPZ COX-2 trwające do 3 lat wykazały zwiększone ryzyko poważnych zakrzepowych zdarzeń sercowo-naczyniowych (CV), w tym zawału mięśnia sercowego (MI) i udaru mózgu, które mogą być śmiertelne. Na podstawie dostępnych danych nie jest jasne, czy ryzyko wystąpienia incydentów zakrzepowych w układzie sercowo-naczyniowym jest podobne w przypadku wszystkich NLPZ. Względny wzrost częstości występowania poważnych incydentów zakrzepowych w układzie sercowo-naczyniowym w porównaniu z wartościami wyjściowymi spowodowany stosowaniem NLPZ wydaje się być podobny u osób z rozpoznaną chorobą sercowo-naczyniową lub czynnikami ryzyka choroby sercowo-naczyniowej lub bez nich. Jednak pacjenci z rozpoznaną chorobą sercowo-naczyniową lub czynnikami ryzyka mieli większą bezwzględną częstość występowania poważnych incydentów zakrzepowych sercowo-naczyniowych ze względu na ich zwiększoną wyjściową częstość występowania. Niektóre badania obserwacyjne wykazały, że to zwiększone ryzyko poważnych incydentów zakrzepowych układu sercowo-naczyniowego rozpoczęło się już w pierwszych tygodniach leczenia. Wzrost ryzyka zakrzepicy sercowo-naczyniowej był najbardziej konsekwentnie obserwowany przy wyższych dawkach.
Aby zminimalizować potencjalne ryzyko wystąpienia niepożądanego zdarzenia sercowo-naczyniowego u pacjentów leczonych NLPZ, należy stosować najniższą skuteczną dawkę przez możliwie najkrótszy czas. Lekarze i pacjenci powinni zachować czujność na rozwój takich zdarzeń przez cały cykl leczenia, nawet przy braku wcześniejszych objawów sercowo-naczyniowych. Pacjentów należy poinformować o objawach poważnych zdarzeń sercowo-naczyniowych oraz o krokach, jakie należy podjąć w przypadku ich wystąpienia.
Nie ma spójnych dowodów na to, że jednoczesne stosowanie aspiryny łagodzi zwiększone ryzyko poważnych incydentów zakrzepicy sercowo-naczyniowej związane ze stosowaniem NLPZ. Jednoczesne stosowanie aspiryny i NLPZ, takich jak diklofenak, zwiększa ryzyko poważnych zdarzeń żołądkowo-jelitowych (GI) (patrz OSTRZEŻENIA ; Krwawienie z przewodu pokarmowego , Owrzodzenie , oraz Perforacja ).
Stan po zabiegu pomostowania tętnic wieńcowych (CABG)
Dwa duże, kontrolowane badania kliniczne NLPZ selektywnego wobec COX-2 w leczeniu bólu w ciągu pierwszych 10 do 14 dni po operacji CABG wykazały zwiększoną częstość występowania zawału mięśnia sercowego i udaru mózgu. NLPZ są przeciwwskazane w CABG (patrz PRZECIWWSKAZANIA ).
Pacjenci po MI
Badania obserwacyjne przeprowadzone w Duńskim Krajowym Rejestrze wykazały, że pacjenci leczeni NLPZ w okresie po MI byli narażeni na zwiększone ryzyko ponownego zawału, zgonów z przyczyn sercowo-naczyniowych i śmiertelności z jakiejkolwiek przyczyny, począwszy od pierwszego tygodnia leczenia. W tej samej kohorcie częstość zgonów w pierwszym roku po MI wynosiła 20 na 100 osobolat u pacjentów leczonych NLPZ w porównaniu z 12 na 100 osobolat u pacjentów nie narażonych na NLPZ. Chociaż bezwzględny wskaźnik zgonów zmniejszył się nieco po pierwszym roku po MI, zwiększone względne ryzyko zgonu u osób stosujących NLPZ utrzymywało się przez co najmniej kolejne cztery lata obserwacji.
Należy unikać stosowania produktu VOLTAREN u pacjentów po niedawno przebytym zawale serca, chyba że spodziewane korzyści przewyższają ryzyko nawrotów zakrzepicy sercowo-naczyniowej. Jeśli VOLTAREN jest stosowany u pacjentów po niedawno przebytym zawale serca, należy monitorować pacjentów pod kątem objawów niedokrwienia serca.
Krwawienie z przewodu pokarmowego, owrzodzenie i perforacja
NLPZ, w tym diklofenak, powodują poważne działania niepożądane ze strony przewodu pokarmowego (GI), w tym zapalenie, krwawienie, owrzodzenie i perforację przełyku, żołądka, jelita cienkiego lub grubego, które mogą być śmiertelne. Te poważne zdarzenia niepożądane mogą wystąpić w dowolnym momencie, z objawami ostrzegawczymi lub bez, u pacjentów leczonych NLPZ. Tylko jeden na pięciu pacjentów, u których wystąpiło poważne zdarzenie niepożądane w górnym odcinku przewodu pokarmowego podczas terapii NLPZ, ma objawy. Owrzodzenia górnego odcinka przewodu pokarmowego, duże krwawienia lub perforacje spowodowane przez NLPZ wystąpiły u około 1% pacjentów leczonych przez 3 do 6 miesięcy i u około 2% do 4% pacjentów leczonych przez jeden rok. Jednak nawet krótkotrwała terapia nie jest pozbawiona ryzyka.
Czynniki ryzyka krwawienia z przewodu pokarmowego, owrzodzeń i perforacji
Pacjenci z chorobą wrzodową i/lub krwawieniem z przewodu pokarmowego stosujący NLPZ mieli ponad 10-krotnie większe ryzyko wystąpienia krwawienia z przewodu pokarmowego w porównaniu z pacjentami bez tych czynników ryzyka. Inne czynniki zwiększające ryzyko krwawienia z przewodu pokarmowego u pacjentów leczonych NLPZ obejmują dłuższy czas leczenia NLPZ, jednoczesne stosowanie doustnych kortykosteroidów, aspiryny, antykoagulantów lub selektywnych inhibitorów wychwytu zwrotnego serotoniny (SSRI); palenie, spożywanie alkoholu, podeszły wiek i zły ogólny stan zdrowia. Większość zgłoszeń po wprowadzeniu do obrotu śmiertelnych zdarzeń ze strony przewodu pokarmowego dotyczyła pacjentów w podeszłym wieku lub osłabionych. Ponadto pacjenci z zaawansowaną chorobą wątroby i/lub koagulopatią są narażeni na zwiększone ryzyko krwawienia z przewodu pokarmowego.
Strategie Zminimalizowania Ryzyka GI u pacjentów leczonych NLPZ
- Użyj najniższej skutecznej dawki przez możliwie najkrótszy czas.
- Unikaj jednoczesnego podawania więcej niż jednego NLPZ
- Należy unikać stosowania u pacjentów z grupy podwyższonego ryzyka, chyba że spodziewane korzyści przewyższają zwiększone ryzyko krwawienia. W przypadku takich pacjentów, a także tych z aktywnym krwawieniem z przewodu pokarmowego, należy rozważyć alternatywne terapie inne niż NLPZ.
- Zachowaj czujność na oznaki i objawy owrzodzenia przewodu pokarmowego i krwawienia podczas leczenia NLPZ.
- W przypadku podejrzenia poważnego zdarzenia niepożądanego ze strony przewodu pokarmowego należy niezwłocznie rozpocząć ocenę i leczenie, a następnie odstawić produkt leczniczy VOLTAREN 50 mg do czasu wykluczenia poważnego zdarzenia niepożądanego ze strony przewodu pokarmowego.
- przypadku jednoczesnego stosowania małej dawki aspiryny w profilaktyce kardiologicznej należy uważniej monitorować pacjentów pod kątem krwawienia z przewodu pokarmowego (patrz ŚRODKI OSTROŻNOŚCI ; Interakcje leków ).
Hepatotoksyczność
W badaniach klinicznych produktów zawierających diklofenak znaczące zwiększenie (tj. ponad 3-krotność górnej granicy normy [GGN]) aminotransferazy asparaginianowej (AspAT) (znanej również jako SGOT) obserwowano u około 2% z około 5700 pacjentów w przez pewien czas podczas leczenia diklofenakiem (nie we wszystkich badaniach mierzono aminotransferazy alaninowej [AlAT]).
dużym, otwartym, kontrolowanym badaniu obejmującym 3700 pacjentów leczonych doustnie diklofenakiem sodowym przez 2 do 6 miesięcy, pacjentów monitorowano najpierw po 8 tygodniach, a 1200 pacjentów monitorowano ponownie po 24 tygodniach. Znaczące podwyższenie ALT i/lub AST wystąpiło u około 4% pacjentów i obejmowało znaczne podwyższenie (ponad 8-krotność GGN) u około 1% z 3700 pacjentów. W tym otwartym badaniu u pacjentów obserwowano częstsze występowanie granicznych (mniej niż 3-krotność GGN), umiarkowanych (3 do 8-krotność GGN) i znacznego (ponad 8-krotność GGN) podwyższenia aktywności AlAT lub AspAT. otrzymujących diklofenak w porównaniu z innymi NLPZ. Zwiększenie aktywności aminotransferaz obserwowano częściej u pacjentów z chorobą zwyrodnieniową stawów niż u pacjentów z reumatoidalnym zapaleniem stawów.
Prawie wszystkie znaczące podwyższenia poziomu transaminaz wykryto, zanim u pacjentów pojawiły się objawy. Nieprawidłowe wyniki badań wystąpiły w ciągu pierwszych 2 miesięcy leczenia diklofenakiem u 42 z 51 pacjentów we wszystkich badaniach, u których wystąpił znaczny wzrost aktywności aminotransferaz.
W raportach po wprowadzeniu leku do obrotu przypadki hepatotoksyczności wywołanej lekiem zgłaszano w pierwszym miesiącu, a w niektórych przypadkach w pierwszych 2 miesiącach leczenia, ale mogą one wystąpić w dowolnym momencie leczenia diklofenakiem. Nadzór po wprowadzeniu do obrotu zgłaszał przypadki ciężkich reakcji wątrobowych, w tym martwicy wątroby, żółtaczki, piorunującego zapalenia wątroby z żółtaczką lub bez oraz niewydolności wątroby. Niektóre z tych zgłoszonych przypadków zakończyły się zgonem lub przeszczepem wątroby.
europejskim retrospektywnym populacyjnym badaniu kliniczno-kontrolnym, 10 przypadków polekowego uszkodzenia wątroby związanego z diklofenakiem przy obecnym stosowaniu w porównaniu z niestosowaniem diklofenaku wiązało się ze statystycznie istotnym 4-krotnym skorygowanym ilorazem szans uszkodzenia wątroby. W tym konkretnym badaniu, w oparciu o ogólną liczbę 10 przypadków uszkodzenia wątroby związanego z diklofenakiem, skorygowany iloraz szans wzrastał dalej wraz z płcią żeńską, dawkami 150 mg lub większymi i czasem stosowania przez ponad 90 dni.
Lekarze powinni oznaczać aktywność aminotransferaz przed rozpoczęciem leczenia oraz okresowo u pacjentów długotrwale leczonych diklofenakiem, ponieważ może rozwinąć się ciężka hepatotoksyczność bez zwiastunów objawów odróżniających. Nie są znane optymalne czasy wykonania pierwszego i kolejnych pomiarów transaminaz. W oparciu o dane z badań klinicznych i doświadczenia po wprowadzeniu produktu do obrotu, transaminazy należy monitorować w ciągu 4 do 8 tygodni po rozpoczęciu leczenia diklofenakiem. Jednak ciężkie reakcje ze strony wątroby mogą wystąpić w dowolnym momencie leczenia diklofenakiem.
Jeśli nieprawidłowe wyniki testów wątrobowych utrzymują się lub pogarszają, jeśli wystąpią objawy kliniczne i (lub) objawy choroby wątroby lub jeśli wystąpią objawy ogólnoustrojowe (np. eozynofilia, wysypka, ból brzucha, biegunka, ciemne zabarwienie moczu itp.), należy natychmiast przerwać stosowanie produktu Voltaren .
Należy poinformować pacjentów o oznakach i objawach ostrzegawczych hepatotoksyczności (np. nudności, zmęczenie, letarg, biegunka, świąd, żółtaczka, tkliwość w prawym górnym kwadrancie i objawy grypopodobne). Jeśli wystąpią objawy kliniczne wskazujące na chorobę wątroby lub jeśli wystąpią objawy ogólnoustrojowe (np. eozynofilia, wysypka itp.), należy natychmiast przerwać podawanie leku VOLTAREN i przeprowadzić ocenę kliniczną pacjenta.
Aby zminimalizować potencjalne ryzyko wystąpienia niepożądanego zdarzenia związanego z wątrobą u pacjentów leczonych preparatem VOLTAREN, należy stosować najniższą skuteczną dawkę przez możliwie najkrótszy czas. Należy zachować ostrożność przepisując VOLTAREN 100 mg jednocześnie z lekami, o których wiadomo, że są potencjalnie hepatotoksyczne (np. paracetamol, antybiotyki, leki przeciwpadaczkowe).
Nadciśnienie
NLPZ, w tym VOLTAREN 100 mg, mogą prowadzić do nowego wystąpienia nadciśnienia lub pogorszenia istniejącego nadciśnienia, co może przyczynić się do zwiększonej częstości występowania incydentów sercowo-naczyniowych. Pacjenci przyjmujący inhibitory konwertazy angiotensyny (ACE), diuretyki tiazydowe lub diuretyki pętlowe mogą mieć osłabioną odpowiedź na te terapie podczas przyjmowania NLPZ (patrz ŚRODKI OSTROŻNOŚCI ; INTERAKCJE Z LEKAMI ).
Monitorować ciśnienie krwi (BP) podczas rozpoczynania leczenia NLPZ oraz w trakcie leczenia.
Niewydolność serca i obrzęk
Metaanaliza randomizowanych badań kontrolowanych Coxib and traditional NSAID Trialists' Collaboration wykazała około 2-krotny wzrost liczby hospitalizacji z powodu niewydolności serca u pacjentów leczonych selektywnie COX-2 i nieselektywnych NLPZ w porównaniu z pacjentami otrzymującymi placebo. W badaniu duńskiego krajowego rejestru pacjentów z niewydolnością serca stosowanie NLPZ zwiększało ryzyko zawału serca, hospitalizacji z powodu niewydolności serca i zgonu.
Ponadto u niektórych pacjentów leczonych NLPZ obserwowano zatrzymanie płynów i obrzęki. Stosowanie diklofenaku może osłabiać działanie na układ sercowo-naczyniowy kilku środków terapeutycznych stosowanych w leczeniu tych schorzeń (np. diuretyków, inhibitorów ACE lub blokerów receptora angiotensyny [ARB]) (patrz INTERAKCJE Z LEKAMI ).
Należy unikać stosowania leku VOLTAREN u pacjentów z ciężką niewydolnością serca, chyba że spodziewane korzyści przewyższają ryzyko pogorszenia niewydolności serca. Jeśli VOLTAREN 50 mg jest stosowany u pacjentów z ciężką niewydolnością serca, należy monitorować pacjentów pod kątem objawów pogorszenia niewydolności serca.
Toksyczność nerek i hiperkaliemia
Toksyczność nerek
Długotrwałe podawanie NLPZ spowodowało martwicę brodawek nerkowych i inne uszkodzenie nerek.
Toksyczny wpływ na nerki obserwowano również u pacjentów, u których prostaglandyny nerkowe pełnią funkcję kompensacyjną w utrzymaniu perfuzji nerek. U tych pacjentów podawanie NLPZ może powodować zależne od dawki zmniejszenie wytwarzania prostaglandyn i, wtórnie, nerkowego przepływu krwi, co może przyspieszyć jawną dekompensację nerek. Największe ryzyko wystąpienia tej reakcji to pacjenci z zaburzeniami czynności nerek, odwodnieniem, hipowolemią, niewydolnością serca, zaburzeniami czynności wątroby, przyjmujący leki moczopędne i inhibitory ACE lub ARB oraz osoby starsze. Po przerwaniu leczenia NLPZ następuje zazwyczaj powrót do stanu sprzed leczenia.
Brak dostępnych informacji z kontrolowanych badań klinicznych dotyczących stosowania preparatu VOLTAREN 100 mg u pacjentów z zaawansowaną chorobą nerek. Działanie leku VOLTAREN 50 mg na nerki może przyspieszyć postęp zaburzeń czynności nerek u pacjentów z istniejącą wcześniej chorobą nerek.
Prawidłowy stan objętości u pacjentów odwodnionych lub z hipowolemią przed rozpoczęciem leczenia preparatem VOLTAREN. Monitorować czynność nerek u pacjentów z zaburzeniami czynności nerek lub wątroby, niewydolnością serca, odwodnieniem lub hipowolemią podczas stosowania leku VOLTAREN (patrz INTERAKCJE Z LEKAMI ). Należy unikać stosowania leku VOLTAREN u pacjentów z zaawansowaną chorobą nerek, chyba że spodziewane korzyści przewyższają ryzyko pogorszenia czynności nerek. Jeśli VOLTAREN 50 mg jest stosowany u pacjentów z zaawansowaną chorobą nerek, należy monitorować pacjentów pod kątem objawów pogorszenia czynności nerek.
Hiperkaliemia
Podczas stosowania NLPZ zgłaszano zwiększenie stężenia potasu w surowicy, w tym hiperkaliemię, nawet u niektórych pacjentów bez zaburzeń czynności nerek. U pacjentów z prawidłową czynnością nerek działania te przypisano stanowi hiporeninemii i hipoaldosteronizmu.
Reakcje anafilaktyczne
Diklofenak był związany z występowaniem reakcji anafilaktycznych u pacjentów z nadwrażliwością na diklofenak lub bez znanej nadwrażliwości na diklofenak oraz u pacjentów z astmą wrażliwą na aspirynę (patrz PRZECIWWSKAZANIA , OSTRZEŻENIA ; Zaostrzenie astmy związanej z wrażliwością na aspirynę ).
Zaostrzenie astmy związane z wrażliwością na aspirynę
Subpopulacja pacjentów z astmą może mieć astmę wrażliwą na aspirynę, która może obejmować przewlekłe zapalenie zatok przynosowych powikłane polipami nosa; ciężki, potencjalnie śmiertelny skurcz oskrzeli; i/lub nietolerancja aspiryny i innych NLPZ. Ze względu na występowanie reakcji krzyżowej między aspiryną i innymi NLPZ u takich pacjentów nadwrażliwych na aspirynę, VOLTAREN 100 mg jest przeciwwskazany u pacjentów z tą postacią wrażliwości na aspirynę (patrz PRZECIWWSKAZANIA ). Gdy VOLTAREN 100 mg jest stosowany u pacjentów z istniejącą wcześniej astmą (bez znanej wrażliwości na aspirynę), należy monitorować pacjentów pod kątem zmian w objawach astmy.
Poważne reakcje skórne
NLPZ, w tym diklofenak, mogą powodować poważne reakcje skórne, takie jak złuszczające zapalenie skóry, zespół Stevensa-Johnsona (SJS) i toksyczna nekroliza naskórka (TEN), które mogą być śmiertelne. Te poważne zdarzenia mogą wystąpić bez ostrzeżenia. Należy poinformować pacjentów o objawach przedmiotowych i podmiotowych ciężkich reakcji skórnych oraz o zaprzestaniu stosowania leku VOLTAREN po pierwszym pojawieniu się wysypki skórnej lub jakichkolwiek innych objawów nadwrażliwości. VOLTAREN jest przeciwwskazany u pacjentów z wcześniejszymi ciężkimi reakcjami skórnymi na NLPZ (patrz PRZECIWWSKAZANIA ).
Reakcja lekowa z eozynofilią i objawami ogólnoustrojowymi (DRESS)
pacjentów przyjmujących NLPZ, takie jak VOLTAREN, zgłaszano reakcje polekowe z eozynofilią i objawami ogólnoustrojowymi (DRESS). Niektóre z tych zdarzeń były śmiertelne lub zagrażały życiu. SUKIENKA zazwyczaj, choć nie wyłącznie, objawia się gorączką, wysypką, powiększeniem węzłów chłonnych i/lub obrzękiem twarzy. Inne objawy kliniczne mogą obejmować zapalenie wątroby, zapalenie nerek, nieprawidłowości hematologiczne, zapalenie mięśnia sercowego lub zapalenie mięśni. Czasami objawy DRESS mogą przypominać ostrą infekcję wirusową. Często występuje eozynofilia. Ponieważ zaburzenie to ma zmienną prezentację, mogą być zaangażowane inne układy narządów nie wymienione tutaj. Należy zauważyć, że wczesne objawy nadwrażliwości, takie jak gorączka lub powiększenie węzłów chłonnych, mogą być obecne, nawet jeśli wysypka nie jest widoczna. Jeśli takie oznaki lub objawy wystąpią, należy przerwać stosowanie leku Voltaren 50 mg i natychmiast ocenić stan pacjenta.
Toksyczność płodu
Przedwczesne zamknięcie przewodu tętniczego płodu
Unikaj stosowania NLPZ, w tym VOLTAREN 50mg, u kobiet w ciąży około 30. tygodnia ciąży i później. NLPZ, w tym VOLTAREN 50 mg, zwiększają ryzyko przedwczesnego zamknięcia przewodu tętniczego płodu mniej więcej w tym wieku ciążowym.
Małowodzie/niewydolność nerek noworodka
Stosowanie NLPZ, w tym preparatu VOLTAREN, w około 20. tygodniu ciąży lub później może powodować zaburzenia czynności nerek płodu prowadzące do małowodzie, a w niektórych przypadkach do zaburzeń czynności nerek u noworodków. Te działania niepożądane obserwuje się średnio po dniach lub tygodniach leczenia, chociaż małowodzie zgłaszano rzadko już po 48 godzinach od rozpoczęcia leczenia NLPZ. Małowodzie jest często, ale nie zawsze, odwracalne po przerwaniu leczenia. Powikłania przedłużonego małowodzia mogą na przykład obejmować przykurcze kończyn i opóźnione dojrzewanie płuc. W niektórych przypadkach zaburzeń czynności nerek noworodków po wprowadzeniu do obrotu konieczne były procedury inwazyjne, takie jak transfuzja wymienna lub dializa.
Jeśli leczenie NLPZ jest konieczne między około 20 tygodniem a 30 tygodniem ciąży, należy ograniczyć stosowanie VOLTAREN do najniższej skutecznej dawki i możliwie najkrótszego czasu trwania. Rozważ monitorowanie ultrasonograficzne płynu owodniowego, jeśli leczenie preparatem VOLTAREN 50 mg trwa dłużej niż 48 godzin. W przypadku wystąpienia małowodzia należy przerwać stosowanie leku VOLTAREN 100 mg i postępować zgodnie z praktyką kliniczną (patrz ŚRODKI OSTROŻNOŚCI ; Ciąża ).
Toksyczność hematologiczna
U pacjentów leczonych NLPZ wystąpiła niedokrwistość. Może to być spowodowane utajoną lub znaczną utratą krwi, zatrzymaniem płynów lub niekompletnie opisanym wpływem na erytropoezę. Jeśli pacjent leczony preparatem VOLTAREN ma jakiekolwiek oznaki lub objawy niedokrwistości, należy monitorować stężenie hemoglobiny lub hematokrytu.
NLPZ, w tym VOLTAREN 100 mg, mogą zwiększać ryzyko krwawienia. Stany współistniejące, takie jak zaburzenia krzepnięcia, jednoczesne stosowanie warfaryny, innych leków przeciwzakrzepowych, leków przeciwpłytkowych (np. aspiryna), inhibitorów wychwytu zwrotnego serotoniny (SSRI) i inhibitorów wychwytu zwrotnego serotoniny i noradrenaliny (SNRI) mogą zwiększać to ryzyko. Należy monitorować tych pacjentów pod kątem oznak krwawienia (patrz INTERAKCJE Z LEKAMI ).
ŚRODKI OSTROŻNOŚCI
Ogólny
Nie można oczekiwać, że VOLTAREN® (tabletki dojelitowe diklofenak sodowy) zastąpi kortykosteroidy lub będzie leczyć niedobór kortykosteroidów. Nagłe odstawienie kortykosteroidów może prowadzić do zaostrzenia choroby. U pacjentów stosujących długotrwałą terapię kortykosteroidami należy powoli zmniejszać terapię, jeśli podjęta zostanie decyzja o odstawieniu kortykosteroidów, a pacjenta należy uważnie obserwować pod kątem jakichkolwiek objawów działań niepożądanych, w tym niewydolności nadnerczy i zaostrzenia objawów zapalenia stawów.
Aktywność farmakologiczna leku VOLTAREN 50 mg w zmniejszaniu gorączki i stanu zapalnego może zmniejszyć użyteczność tych objawów diagnostycznych w wykrywaniu powikłań przypuszczalnie niezakaźnych, bolesnych stanów.

Informacje dla pacjentów
Poinformuj pacjenta, aby przeczytał zatwierdzone przez FDA oznakowanie pacjenta ( Przewodnik po lekach ), która towarzyszy każdej zrealizowanej recepcie. Należy poinformować pacjentów, rodziny lub ich opiekunów o następujących informacjach przed rozpoczęciem leczenia preparatem VOLTAREN oraz okresowo w trakcie trwającej terapii.
Zdarzenia zakrzepowo-sercowe
Należy doradzić pacjentom, aby zwracali uwagę na objawy zakrzepicy sercowo-naczyniowej, w tym ból w klatce piersiowej, duszność, osłabienie lub niewyraźną mowę, i natychmiast zgłaszali każdy z tych objawów swojemu lekarzowi (patrz OSTRZEŻENIA ; Zdarzenia zakrzepowo-sercowe ).
Krwawienie z przewodu pokarmowego, owrzodzenie i perforacja
Poradź pacjentom, aby zgłaszali swojemu lekarzowi objawy owrzodzeń i krwawień, w tym ból w nadbrzuszu, niestrawność, smoliste stolce i krwawe wymioty. W przypadku jednoczesnego stosowania kwasu acetylosalicylowego w małych dawkach w profilaktyce kardiologicznej należy poinformować pacjentów o zwiększonym ryzyku wystąpienia objawów przedmiotowych i podmiotowych krwawienia z przewodu pokarmowego (patrz OSTRZEŻENIE ; Krwawienie z przewodu pokarmowego , Owrzodzenie , oraz Perforacja ).
Hepatotoksyczność
Należy poinformować pacjentów o oznakach i objawach ostrzegawczych hepatotoksyczności (np. nudności, zmęczenie, letarg, świąd, biegunka, żółtaczka, tkliwość w prawym górnym kwadrancie i objawy grypopodobne). W takim przypadku należy poinstruować pacjentów, aby odstawili produkt leczniczy Voltaren 50 mg i zgłosili się do natychmiastowego leczenia (patrz OSTRZEŻENIA ; Hepatotoksyczność ).
Niewydolność serca i obrzęk
Należy doradzić pacjentom, aby zwracali uwagę na objawy zastoinowej niewydolności serca, w tym duszność, niewyjaśniony przyrost masy ciała lub obrzęki, i skontaktowali się z lekarzem, jeśli takie objawy wystąpią (patrz OSTRZEŻENIA ; Niewydolność serca i obrzęk ).
Reakcje anafilaktyczne
Poinformuj pacjentów o objawach reakcji anafilaktycznej (np. trudności w oddychaniu, obrzęk twarzy lub gardła). Należy poinstruować pacjentów, aby w razie ich wystąpienia szukali natychmiastowej pomocy w nagłych wypadkach (patrz OSTRZEŻENIA ; Reakcje anafilaktyczne ).
Poważne reakcje skórne, w tym SUKIENKA
Należy doradzić pacjentom natychmiastowe przerwanie stosowania leku Voltaren 50 mg, jeśli wystąpi u nich jakakolwiek wysypka lub gorączka, i jak najszybciej skontaktować się z lekarzem (patrz OSTRZEŻENIA ; Poważne reakcje skórne ).
Płodność kobiet
Poinformuj kobiety w wieku rozrodczym, które pragną zajść w ciążę, że NLPZ, w tym VOLTAREN 50 mg, mogą być związane z odwracalnym opóźnieniem owulacji (patrz ŚRODKI OSTROŻNOŚCI ; Karcynogeneza, mutageneza, upośledzenie płodności ).
Toksyczność płodu
Należy poinformować kobiety w ciąży, aby unikały stosowania leku Voltaren i innych NLPZ, począwszy od 30 tygodnia ciąży, ze względu na ryzyko przedwczesnego zamknięcia przewodu tętniczego płodu. Jeśli leczenie preparatem VOLTAREN jest konieczne u kobiety w ciąży między około 20. a 30. tygodniem ciąży, należy jej poinformować, że może być konieczne monitorowanie jej pod kątem małowodzia, jeśli leczenie trwa dłużej niż 48 godzin. (Widzieć OSTRZEŻENIA ; Toksyczność płodu , ŚRODKI OSTROŻNOŚCI , Ciąża ).
Unikaj jednoczesnego stosowania NLPZ
Należy poinformować pacjentów, że jednoczesne stosowanie produktu leczniczego VOLTAREN 50 mg z innymi NLPZ lub salicylanami (np. diflunisalem, salsalatem) nie jest zalecane ze względu na zwiększone ryzyko toksyczności żołądkowo-jelitowej oraz niewielki lub żaden wzrost skuteczności (patrz OSTRZEŻENIA ; Krwawienie z przewodu pokarmowego , Owrzodzenie , oraz Perforacja i interakcje leków ). Ostrzegaj pacjentów, że NLPZ mogą być obecne w lekach „bez recepty” stosowanych w leczeniu przeziębienia, gorączki lub bezsenności.
Stosowanie NLPZ i aspiryny w niskich dawkach
Należy poinformować pacjentów, aby nie stosowali małych dawek aspiryny jednocześnie z preparatem VOLTAREN, dopóki nie porozmawiają z lekarzem (patrz INTERAKCJE Z LEKAMI ).
Maskowanie stanu zapalnego i gorączki
Aktywność farmakologiczna preparatu VOLTAREN w zmniejszaniu stanu zapalnego i ewentualnie gorączki może zmniejszyć użyteczność objawów diagnostycznych w wykrywaniu zakażeń.
Monitoring laboratoryjny
Ponieważ poważne krwawienie z przewodu pokarmowego, hepatotoksyczność i uszkodzenie nerek mogą wystąpić bez objawów lub oznak ostrzegawczych, należy rozważyć okresowe monitorowanie pacjentów długotrwale leczonych NLPZ za pomocą CBC i profilu chemicznego (patrz OSTRZEŻENIA ; Krwawienie z przewodu pokarmowego , Owrzodzenie i perforacja , oraz Hepatotoksyczność ).
rakotwórczość, mutageneza, upośledzenie płodności
Karcynogeneza
Długotrwałe badania rakotwórczości na szczurach, którym podawano diklofenak sodowy w dawce do 2 mg/kg/dobę (około 0,1-krotność maksymalnej zalecanej dawki u ludzi (MRHD) VOLTAREN 50 mg, 200 mg/dobę, na podstawie porównania powierzchni ciała (BSA) brak znaczącego wzrostu zachorowalności na nowotwory. Dwuletnie badanie rakotwórczości przeprowadzone na myszach stosujących diklofenak sodu w dawkach do 0,3 mg/kg/dzień (około 0,007-krotności MRHD na podstawie porównania BSA) u samców i 1 mg/kg/dobę (około 0,02-krotność MRHD na podstawie porównania BSA) Porównanie BSA) u kobiet nie wykazało żadnego potencjału onkogennego.
Mutageneza
Diklofenak sodowy nie wykazywał działania mutagennego w testach mutacji punktowych in vitro w układach testowych ssaków (chłoniak myszy) i drobnoustrojów (drożdże, Ames) i nie wykazywał działania mutagennego w kilku badaniach in vitro i in vivo na ssakach, w tym w badaniach chromosomalnych nabłonka zarodków samców i nabłonka germinalnego u myszy oraz badania anomalii jądra i aberracji chromosomowych u chomików chińskich.
Upośledzenie płodności
Diklofenak sodowy podawany samcom i samicom szczurów w dawce 4 mg/kg/dobę (około 0,2-krotność MRHD na podstawie porównania BSA) nie wpływał na płodność.
W oparciu o mechanizm działania, stosowanie NLPZ, w których pośredniczą prostaglandyny, w tym VOLTAREN 100 mg, może opóźnić lub zapobiec pęknięciu pęcherzyków jajnikowych, które u niektórych kobiet jest związane z odwracalną niepłodnością. Opublikowane badania na zwierzętach wykazały, że podawanie inhibitorów syntezy prostaglandyn może zaburzać pośredniczone przez prostaglandyny pęknięcie pęcherzyka wymagane do owulacji. Niewielkie badania u kobiet leczonych NLPZ wykazały również odwracalne opóźnienie owulacji. Rozważ odstawienie NLPZ, w tym VOLTAREN 100 mg, u kobiet, które mają trudności z zajściem w ciążę lub są w trakcie badania niepłodności.
Ciąża
Podsumowanie ryzyka
Stosowanie NLPZ, w tym VOLTAREN, może spowodować przedwczesne zamknięcie przewodu tętniczego płodu i zaburzenia czynności nerek płodu prowadzące do małowodzie, a w niektórych przypadkach do niewydolności nerek u noworodków. Ze względu na te zagrożenia należy ograniczyć dawkę i czas stosowania leku VOLTAREN między około 20 a 30 tygodniem ciąży oraz unikać stosowania leku VOLTAREN około 30 tygodnia ciąży i później (patrz OSTRZEŻENIA ; Toksyczność płodu ).
Przedwczesne zamknięcie przewodu tętniczego płodu
Stosowanie NLPZ, w tym VOLTAREN 100 mg, około 30 tygodnia ciąży lub później zwiększa ryzyko przedwczesnego zamknięcia przewodu tętniczego płodu.
Małowodzie/niewydolność nerek noworodka
Stosowanie NLPZ w około 20. tygodniu ciąży lub później w ciąży wiązało się z przypadkami dysfunkcji nerek płodu prowadzącej do małowodzia, a w niektórych przypadkach do niewydolności nerek u noworodków.
Nie ma odpowiednich i dobrze kontrolowanych badań produktu VOLTAREN 100 mg u kobiet w ciąży.
Dane z badań obserwacyjnych dotyczące potencjalnego ryzyka dla zarodka i płodu związanego ze stosowaniem NLPZ u kobiet w pierwszym lub drugim trymestrze ciąży są niejednoznaczne.
badaniach na zwierzętach dotyczących reprodukcji nie zaobserwowano dowodów działania teratogennego u myszy, szczurów lub królików, którym podawano diklofenak w okresie organogenezy w dawkach do około 0,5, 0,5 i 1 razy większej od maksymalnej zalecanej dawki u ludzi (MRHD) produktu VOLTAREN 200 mg/dobę, pomimo toksycznego wpływu tych dawek na matkę i płód (patrz Dane ).
Na podstawie opublikowanych danych na zwierzętach wykazano, że prostaglandyny odgrywają ważną rolę w przepuszczalności naczyń endometrium, implantacji blastocysty i decydualizacji. W badaniach na zwierzętach podawanie inhibitorów syntezy prostaglandyn, takich jak diklofenak, powodowało zwiększenie poronień przed i po implantacji. Wykazano również, że prostaglandyny odgrywają ważną rolę w rozwoju nerek płodu. W opublikowanych badaniach na zwierzętach donoszono, że inhibitory syntezy prostaglandyn, podawane w klinicznie istotnych dawkach, zaburzają rozwój nerek.
Szacowane ryzyko tła poważnych wad wrodzonych i poronienia dla wskazanej populacji jest nieznane. Wszystkie ciąże wiążą się z ryzykiem wady wrodzonej, utraty lub innych niekorzystnych skutków. W ogólnej populacji Stanów Zjednoczonych szacowane podstawowe ryzyko poważnych wad wrodzonych i poronienia w klinicznie rozpoznanych ciążach wynosi odpowiednio 2% do 4% i 15% do 20%.
Rozważania kliniczne
Działania niepożądane u płodu/noworodka
Przedwczesne zamknięcie przewodu tętniczego płodu:
Należy unikać stosowania NLPZ u kobiet w około 30. tygodniu ciąży i później w ciąży, ponieważ NLPZ, w tym VOLTAREN, mogą spowodować przedwczesne zamknięcie przewodu tętniczego płodu (patrz OSTRZEŻENIA ; Toksyczność płodu ).
Małowodzie/niewydolność nerek noworodka:
Jeśli NLPZ jest konieczne w około 20. tygodniu ciąży lub później, ogranicz stosowanie do najniższej skutecznej dawki i możliwie najkrótszego czasu trwania. Jeśli leczenie preparatem VOLTAREN 50 mg trwa dłużej niż 48 godzin, należy rozważyć monitorowanie za pomocą ultrasonografii pod kątem małowodzia. W przypadku wystąpienia małowodzia należy odstawić VOLTAREN i postępować zgodnie z praktyką kliniczną (patrz OSTRZEŻENIA ; Toksyczność płodu ).
Dane
Dane ludzkie
Przedwczesne zamknięcie przewodu tętniczego płodu:
Z opublikowanego piśmiennictwa wynika, że stosowanie NLPZ w około 30. tygodniu ciąży i później w ciąży może spowodować przedwczesne zamknięcie przewodu tętniczego płodu.
Małowodzie/niewydolność nerek noworodka:
Opublikowane badania i doniesienia po wprowadzeniu do obrotu opisują stosowanie NLPZ przez matkę w około 20. tygodniu ciąży lub później w związku z zaburzeniami czynności nerek płodu prowadzącymi do małowodzia, a w niektórych przypadkach z zaburzeniami czynności nerek u noworodków. Te działania niepożądane obserwuje się średnio po dniach lub tygodniach leczenia, chociaż małowodzie zgłaszano rzadko już po 48 godzinach od rozpoczęcia leczenia NLPZ. W wielu przypadkach, ale nie we wszystkich, spadek płynu owodniowego był przemijający i odwracalny wraz z odstawieniem leku. Istnieje ograniczona liczba opisów przypadków stosowania NLPZ u matek i zaburzeń czynności nerek u noworodków bez małowodzia, z których niektóre były nieodwracalne. Niektóre przypadki dysfunkcji nerek noworodków wymagały leczenia za pomocą procedur inwazyjnych, takich jak transfuzja wymienna lub dializa.
Ograniczenia metodologiczne tych badań i raportów postmarketingowych obejmują brak grupy kontrolnej; ograniczone informacje dotyczące dawki, czasu trwania i czasu ekspozycji na lek; i jednoczesne stosowanie innych leków. Ograniczenia te uniemożliwiają ustalenie wiarygodnego oszacowania ryzyka niekorzystnych wyników dla płodu i noworodka przy stosowaniu NLPZ u matki. Ponieważ opublikowane dane dotyczące bezpieczeństwa u noworodków dotyczyły głównie wcześniaków, możliwość uogólnienia niektórych zgłoszonych zagrożeń dla urodzonego w terminie niemowlęcia narażonego na NLPZ poprzez stosowanie przez matkę jest niepewna.
Dane zwierząt
Badania reprodukcyjne i rozwojowe na zwierzętach wykazały, że podawanie diklofenaku sodu podczas organogenezy nie powodowało działania teratogennego pomimo indukcji toksyczności u matki i płodu u myszy przy dawkach doustnych do 20 mg/kg mc./dobę (około 0,5-krotności maksymalnej zalecanej dawki u ludzi [MRHD ] VOLTAREN, 200 mg/dobę, na podstawie porównania powierzchni ciała (BSA)), a u szczurów i królików w dawkach doustnych do 10 mg/kg/dobę (odpowiednio około 0,5 i 1 razy MRHD na podstawie BSA porównanie). W badaniu, w którym ciężarnym szczurom podawano doustnie diklofenak w dawce 2 lub 4 mg/kg (0,1 i 0,2-krotność MRHD na podstawie BSA) od 15. dnia ciąży do 21. dnia laktacji, odnotowano znaczną toksyczność matczyną (zapalenie otrzewnej, śmiertelność). Te dawki toksyczne dla matki były związane z dystocją, przedłużeniem ciąży, zmniejszeniem masy i wzrostu płodu oraz zmniejszoną przeżywalnością płodu. Wykazano, że diklofenak przenika przez barierę łożyskową u myszy, szczurów i ludzi.
Praca lub dostawa
Nie przeprowadzono badań dotyczących wpływu leku VOLTAREN podczas porodu lub porodu. W badaniach na zwierzętach NLPZ, w tym diklofenak, hamują syntezę prostaglandyn, powodują opóźniony poród i zwiększają częstość martwych urodzeń.
Matki karmiące
Podsumowanie ryzyka
Na podstawie dostępnych danych diklofenak może być obecny w mleku ludzkim. Należy wziąć pod uwagę korzyści rozwojowe i zdrowotne wynikające z karmienia piersią, a także kliniczne zapotrzebowanie matki na VOLTAREN i wszelkie potencjalne niekorzystne skutki dla karmionego piersią dziecka z VOLTAREN 100 mg lub z podstawowego stanu matki.
Dane
Jedna kobieta leczona doustnie solą diklofenaku, 150 mg/dzień, miała poziom diklofenaku w mleku 100 mcg/l, co odpowiada dawce dla niemowląt wynoszącej około 0,03 mg/kg/dzień. Diklofenak był niewykrywalny w mleku kobiecym u 12 kobiet stosujących diklofenak (po 100 mg/dobę doustnie przez 7 dni lub po pojedynczej dawce 50 mg podanej domięśniowo bezpośrednio po porodzie).
Zastosowanie pediatryczne
Bezpieczeństwo i skuteczność u pacjentów pediatrycznych nie zostały ustalone.
Zastosowanie geriatryczne
Pacjenci w podeszłym wieku, w porównaniu z młodszymi pacjentami, są narażeni na większe ryzyko poważnych działań niepożądanych ze strony układu krążenia, przewodu pokarmowego i (lub) nerek związanych z NLPZ. Jeśli przewidywane korzyści dla pacjenta w podeszłym wieku przewyższają potencjalne ryzyko, należy rozpocząć dawkowanie od dolnej granicy zakresu dawkowania i monitorować pacjentów pod kątem działań niepożądanych (patrz OSTRZEŻENIA ; Zdarzenia zakrzepowo-sercowe , Przewód pokarmowy Krwawienie, owrzodzenie i perforacja, hepatotoksyczność, toksyczność nerek i hiperkaliemia, ŚRODKI OSTROŻNOŚCI; Monitoring laboratoryjny).
Wiadomo, że diklofenak jest w znacznym stopniu wydalany przez nerki, a ryzyko działań niepożądanych tego leku może być większe u pacjentów z zaburzeniami czynności nerek. Ponieważ u pacjentów w podeszłym wieku istnieje większe prawdopodobieństwo upośledzenia czynności nerek, należy zachować ostrożność przy doborze dawki i może być przydatne monitorowanie czynności nerek (patrz FARMAKOLOGIA KLINICZNA , DZIAŁANIA NIEPOŻĄDANE ).
PRZEDAWKOWAĆ
Nie podano informacji
PRZECIWWSKAZANIA
VOLTAREN® jest przeciwwskazany u następujących pacjentów:
- Znana nadwrażliwość (np. reakcje anafilaktyczne i poważne reakcje skórne) na diklofenak lub którykolwiek składnik produktu leczniczego (patrz OSTRZEŻENIA ; Reakcje anafilaktyczne , Poważne reakcje skórne ).
- Historia astmy, pokrzywki lub innych reakcji alergicznych po przyjęciu aspiryny lub innych NLPZ. U takich pacjentów zgłaszano ciężkie, czasami prowadzące do zgonu reakcje anafilaktyczne na NLPZ (patrz OSTRZEŻENIA ; Reakcja anafilaktyczna , Zaostrzenie astmy związanej z wrażliwością na aspirynę ).
- W przypadku operacji pomostowania aortalno-wieńcowego (CABG) (patrz OSTRZEŻENIA ; Zdarzenia zakrzepowo-sercowe ).
FARMAKOLOGIA KLINICZNA
Mechanizm akcji
Diklofenak ma właściwości przeciwbólowe, przeciwzapalne i przeciwgorączkowe.
Mechanizm działania VOLTAREN 100 mg, podobnie jak innych NLPZ, nie jest w pełni poznany, ale obejmuje hamowanie cyklooksygenazy (COX-1 i COX-2).
Diklofenak jest silnym inhibitorem syntezy prostaglandyn in vitro. Stężenia diklofenaku osiągane podczas terapii wywoływały efekty in vivo. Prostaglandyny uwrażliwiają nerwy doprowadzające i nasilają działanie bradykininy w wywoływaniu bólu w modelach zwierzęcych. Prostaglandyny są mediatorami stanu zapalnego. Ponieważ diklofenak jest inhibitorem syntezy prostaglandyn, jego mechanizm działania może wynikać ze zmniejszenia stężenia prostaglandyn w tkankach obwodowych.
Farmakokinetyka
Wchłanianie
Diklofenak jest wchłaniany w 100% po podaniu doustnym w porównaniu z podaniem dożylnym (IV), co mierzy się na podstawie odzysku moczu. Jednak ze względu na metabolizm pierwszego przejścia tylko około 50% wchłoniętej dawki jest dostępne ogólnoustrojowo (patrz Tabela 1). Pokarm nie ma znaczącego wpływu na stopień wchłaniania diklofenaku. Jednak zwykle występuje opóźnienie w rozpoczęciu wchłaniania o 1 do 4,5 godziny i zmniejszenie maksymalnego stężenia w osoczu o mniej niż 20%.
Dystrybucja
Pozorna objętość dystrybucji (V/F) diklofenaku sodu wynosi 1,4 l/kg.
Diklofenak wiąże się w ponad 99% z białkami ludzkiej surowicy, głównie z albuminą. Wiązanie z białkami surowicy jest stałe w zakresie stężeń (0,15 do 105 µg/ml) osiąganym przy zalecanych dawkach.
Diklofenak dyfunduje do i z płynu maziowego. Dyfuzja do stawu występuje, gdy poziomy w osoczu są wyższe niż w mazi stawowej, po czym proces się odwraca, a poziomy mazi stawowej są wyższe niż poziomy w osoczu. Nie wiadomo, czy dyfuzja do stawu odgrywa rolę w skuteczności diklofenaku.
Eliminacja
Metabolizm
ludzkim osoczu i moczu zidentyfikowano pięć metabolitów diklofenaku. Metabolity obejmują 4'hydroksy-, 5-hydroksy-, 3'-hydroksy-, 4',5-dihydroksy- i 3'-hydroksy-4'-metoksy-diklofenak. Główny metabolit diklofenaku, 4'-hydroksy-diklofenak, ma bardzo słabą aktywność farmakologiczną. W tworzeniu 4'hydroksydiklofenaku pośredniczy głównie CYP2C9. Zarówno diklofenak, jak i jego oksydacyjne metabolity ulegają glukuronidacji lub siarczanowaniu, a następnie wydalaniu z żółcią. Glukuronidacja acylowa za pośrednictwem UGT2B7 i utlenianie za pośrednictwem CYP2C8 mogą również odgrywać rolę w metabolizmie diklofenaku. CYP3A4 odpowiada za powstawanie mniejszych metabolitów, 5-hydroksy- i 3'-hydroksy-diklofenaku. U pacjentów z zaburzeniami czynności nerek maksymalne stężenia metabolitów 4'-hydroksy- i 5-hydroksy-diklofenaku wynosiły około 50% i 4% związku macierzystego po pojedynczej dawce doustnej w porównaniu z 27% i 1% u zdrowych osób.
Wydalanie
Diklofenak jest eliminowany poprzez metabolizm, a następnie wydalanie glukuronidu i koniugatów siarczanowych metabolitów z moczem i żółcią. Niewielki lub żaden niezmieniony diklofenak jest wydalany z moczem. Około 65% dawki jest wydalane z moczem, a około 35% z żółcią w postaci koniugatów niezmienionego diklofenaku z metabolitami. Ponieważ wydalanie nerkowe nie jest istotną drogą eliminacji niezmienionego diklofenaku, nie jest konieczne dostosowanie dawkowania u pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności nerek. Końcowy okres półtrwania niezmienionego diklofenaku wynosi około 2 godziny.
Specjalne populacje
Pediatryczny
Nie badano farmakokinetyki produktu VOLTAREN u dzieci.
Wyścig
Różnice farmakokinetyczne ze względu na rasę nie zostały zidentyfikowane.
Niewydolność wątroby
Metabolizm wątrobowy odpowiada za prawie 100% wydalania preparatu VOLTAREN w dawce 100 mg, dlatego pacjenci z chorobą wątroby mogą wymagać zmniejszonych dawek preparatu VOLTAREN 50 mg w porównaniu z pacjentami z prawidłową czynnością wątroby.
Zaburzenia czynności nerek
Farmakokinetykę diklofenaku badano u osób z niewydolnością nerek. W badaniach z udziałem pacjentów z zaburzeniami czynności nerek nie wykryto różnic w farmakokinetyce diklofenaku. U pacjentów z zaburzeniami czynności nerek (klirens inuliny 60 do 90, 30 do 60 i mniej niż 30 ml/min; N = 6 w każdej grupie), wartości pola pod krzywą (AUC) i szybkość eliminacji były porównywalne z wartościami u zdrowych osób. .
Badania interakcji leków
Worykonazol
W przypadku jednoczesnego podawania z worykonazolem (inhibitor enzymów CYP2C9, 2C19 i 3A4), Cmax i AUC diklofenaku zwiększyły się odpowiednio o 114% i 78% (patrz INTERAKCJE Z LEKAMI ).
Aspiryna
Gdy NLPZ podawano z aspiryną, wiązanie NLPZ z białkami było zmniejszone, chociaż klirens wolnego NLPZ nie uległ zmianie. Kliniczne znaczenie tej interakcji nie jest znane. W Tabeli 2 przedstawiono klinicznie istotne interakcje NLPZ z aspiryną (patrz INTERAKCJE Z LEKAMI ).
INFORMACJA O PACJENCIE
Przewodnik po lekach
VOLTAREN® (Vowl-tar-uhn) (diklofenak sodowy) tabletki powlekane
Jakie są najważniejsze informacje, które powinienem wiedzieć o lekach zwanych niesteroidowymi lekami przeciwzapalnymi (NLPZ)?
NLPZ mogą powodować poważne skutki uboczne, w tym:
- Zwiększone ryzyko zawału serca lub udaru mózgu, które mogą prowadzić do śmierci. Ryzyko to może wystąpić na początku leczenia i może wzrosnąć:
- ze wzrastającymi dawkami NLPZ
- przy dłuższym stosowaniu NLPZ
Nie należy przyjmować NLPZ bezpośrednio przed lub po operacji serca zwanej „pomostowaniem aortalno-wieńcowym (CABG).” Unikaj przyjmowania NLPZ po niedawnym zawale serca, chyba że zaleci Ci to Twój lekarz. Możesz mieć zwiększone ryzyko kolejnego zawału serca, jeśli przyjmujesz NLPZ po niedawnym zawale serca.
- Zwiększone ryzyko krwawienia, owrzodzeń i pęknięć (perforacji) przełyku (przewodu prowadzącego z jamy ustnej do żołądka), żołądka i jelit:
- w dowolnym momencie podczas użytkowania
- bez objawów ostrzegawczych
- które mogą spowodować śmierć
Ryzyko owrzodzenia lub krwawienia wzrasta wraz z:
- wrzody żołądka w przeszłości lub krwawienie z żołądka lub jelit po zastosowaniu NLPZ
- przyjmowanie leków zwanych „kortykosteroidami”, „antykoagulantami”, „SSRI” lub „SNRI”
- rosnące dawki NLPZ
- dłuższe stosowanie NLPZ
- palenie
- picie alkoholu
- starszy wiek
- słabe zdrowie
- zaawansowana choroba wątroby
- problemy z krwawieniem
NLPZ powinny być stosowane wyłącznie:
- dokładnie zgodnie z zaleceniami
- w najniższej możliwej dawce dla twojego leczenia
- na jak najkrótszy czas
Co to są NLPZ?
NLPZ są stosowane w leczeniu bólu i zaczerwienienia, obrzęku i gorąca (zapalenia) z powodu schorzeń, takich jak różne rodzaje zapalenia stawów, skurcze menstruacyjne i inne rodzaje krótkotrwałego bólu.
Kto nie powinien brać NLPZ?
Nie przyjmuj NLPZ:
- jeśli miałeś atak astmy, pokrzywkę lub inną reakcję alergiczną na aspirynę lub inne NLPZ.
- tuż przed lub po operacji pomostowania aortalno-wieńcowego.
Przed zażyciem NLPZ poinformuj swojego lekarza o wszystkich swoich schorzeniach, w tym jeśli:
- masz problemy z wątrobą lub nerkami
- mieć wysokie ciśnienie krwi
- mieć astmę
- są w ciąży lub planują zajść w ciążę. Przyjmowanie NLPZ w około 20. tygodniu ciąży lub później może zaszkodzić nienarodzonemu dziecku. Jeśli musisz zażywać NLPZ przez ponad 2 dni między 20. a 30. tygodniem ciąży, Twój lekarz może być zmuszony do monitorowania ilości płynu w macicy wokół Twojego dziecka. Nie należy przyjmować NLPZ po około 30 tygodniach ciąży.
- karmisz piersią lub planujesz karmić piersią.
Poinformuj swojego lekarza o wszystkich przyjmowanych lekach, w tym o lekach na receptę lub dostępnych bez recepty, witaminach lub suplementach ziołowych. NLPZ i niektóre inne leki mogą wchodzić ze sobą w interakcje i powodować poważne skutki uboczne. Nie rozpoczynaj przyjmowania nowych leków bez uprzedniej konsultacji z lekarzem.
Jakie są możliwe skutki uboczne NLPZ?
NLPZ mogą powodować poważne skutki uboczne, w tym:
Zobacz „Jakie są najważniejsze informacje, które powinienem wiedzieć o lekach zwanych niesteroidowymi lekami przeciwzapalnymi (NLPZ)?”
- nowe lub gorsze wysokie ciśnienie krwi
- niewydolność serca
- problemy z wątrobą, w tym niewydolność wątroby
- problemy z nerkami, w tym niewydolność nerek
- niski poziom czerwonych krwinek (niedokrwistość)
- zagrażające życiu reakcje skórne
- zagrażające życiu reakcje alergiczne
- Inne skutki uboczne NLPZ obejmują: ból brzucha, zaparcia, biegunka, gazy, zgaga, nudności, wymioty, zawroty głowy
Natychmiast uzyskaj pomoc w nagłych wypadkach, jeśli wystąpi którykolwiek z następujących objawów:
- duszność lub trudności w oddychaniu
- ból w klatce piersiowej
- osłabienie w jednej części lub boku ciała
- bełkotliwa wymowa
- obrzęk twarzy lub gardła
Przestań brać NLPZ i natychmiast skontaktuj się z lekarzem, jeśli wystąpi którykolwiek z następujących objawów:
- mdłości
- krew wymiotów
- bardziej zmęczony lub słabszy niż zwykle
- biegunka
- swędzący
- w wypróżnieniach pojawia się krew lub jest czarna i lepka jak smoła
- Twoja skóra lub oczy wyglądają na żółte
- niestrawność lub ból brzucha
- objawy grypopodobne
- nietypowy przyrost masy ciała
- wysypka skórna lub pęcherze z gorączką
- obrzęk rąk i nóg, dłoni i stóp
Jeśli zażyjesz zbyt dużo NLPZ, skontaktuj się z lekarzem lub natychmiast uzyskaj pomoc medyczną.
To nie wszystkie możliwe skutki uboczne NLPZ. Aby uzyskać więcej informacji, zapytaj swojego lekarza lub farmaceutę o NLPZ. Zadzwoń do lekarza, aby uzyskać poradę medyczną na temat skutków ubocznych. Możesz zgłosić skutki uboczne do FDA pod numerem 1-800-FDA1088
Inne informacje o NLPZ.
- Aspiryna jest lekiem NLPZ, ale nie zwiększa ryzyka zawału serca. Aspiryna może powodować krwawienie w mózgu, żołądku i jelitach. Aspiryna może również powodować wrzody żołądka i jelit.
- Niektóre NLPZ są sprzedawane w mniejszych dawkach bez recepty (bez recepty). Porozmawiaj ze swoim lekarzem przed użyciem NLPZ dostępnych bez recepty przez ponad 10 dni.
Ogólne informacje o bezpiecznym i skutecznym stosowaniu NLPZ.
Leki są czasami przepisywane do celów innych niż wymienione w Przewodniku po lekach. Nie należy stosować NLPZ w stanie, na który nie został przepisany. Nie podawaj NLPZ innym osobom, nawet jeśli mają takie same objawy jak ty. Może im to zaszkodzić.
Jeśli chcesz uzyskać więcej informacji na temat NLPZ, porozmawiaj ze swoim lekarzem. Możesz poprosić farmaceutę lub pracownika służby zdrowia o informacje na temat NLPZ, które są przeznaczone dla pracowników służby zdrowia.
Ten przewodnik po lekach został zatwierdzony przez amerykańską Agencję ds. Żywności i Leków. Poprawiono: kwiecień 2021